Introducción
La habilidad para definir resultados y, en consecuencia, diseñar abordajes para el manejo de enfermedades malignas raras, es extremadamente difícil. El sarcoma de tejidos blandos es un grupo de diversas enfermedades malignas con presentación, comportamiento y resultado variables. Los sarcomas de tejidos blandos son complicados por el hecho de que representan, al menos, 80 tipos y subtipos histológicos potencialmente malignos; de ellos, el 50% son completamente malignos y comúnmente metastatizan, y el 50% son agresivos localmente, con algún potencial limitado de metástasis. Se considera que la mayoría de los sarcomas de tejidos blandos son esporádicos; sin embargo, en una pequeña proporción de los casos, factores genéticos, linfedema y terapia radiante previa, han sido identificados como factores predisponentes o asociados. Los sarcomas muestran varios grados de diferenciación mesenquimática, haciendo difícil el diagnóstico y la predicción de su comportamiento [1]. Ese problema se agrava por la naturaleza de estas enfermedades, algunas con progresión temprana, recurrencia y fallecimiento, y otras que continúan con recidivas múltiples o tardías por décadas.
Las bases de datos prospectivas, diseñadas cuidadosamente para recolectar variables importantes de los pacientes, predisposición, tumor y tratamientos, constituyen un abordaje que permite la definición de variables pronósticas para el resultado, brindando una descripción de la naturaleza de la recidiva y de la biología y, sin son seguidas asiduamente durante décadas, constituyen una rica fuente de atención, basada en conocimiento. En este reporte, los autores se enfocan en una serie grande de pacientes, recolectada prospectivamente en una única institución, ingresados y seguidos durante 3 décadas.
Materiales y métodos
En julio de 1982, se comenzó una base de datos prospectiva de pacientes internados, para todos los pacientes adultos (mayores de 16 años) admitidos en la institución, para el manejo quirúrgico de sarcomas de tejidos blandos. Eso incluye a pacientes presentando enfermedad primaria, localmente recidivada o metastásica, si iban a ser sometidos a un procedimiento quirúrgico. Los pacientes que fueron vistos en consulta o los que nunca fueron admitidos para un evento quirúrgico, no fueron incluidos. Esa fue una decisión deliberada, por la dificultad para el seguimiento de una multitud de pacientes derivados para una segunda opinión y/o tratamiento ambulatorio.
La decisión se tomó para incluir a los pacientes internados sometidos a procedimientos quirúrgicos, de manera de tener una rápida disponibilidad de tejidos para una clara definición del tipo y subtipo histológicos. Se registró prospectivamente una cuidadosa clasificación de otras característicos de los pacientes, del tumor y del tratamiento. Además, se pudo obtener, al momento de la presentación, un consentimiento para la recolección no sólo de tejido tumoral, sino también del tejido normal adyacente y, en años posteriores, de sangre, para el análisis de la línea germinal. Los datos son ingresados y revisados por el equipo de manejo de la enfermedad sarcomatosa, al momento de la admisión del paciente para internación, inicialmente semanalmente y después bisemanalmente, con todas las muestras de tumor clasificadas por tipo histológico, subtipo y grado (bajo vs alto) por un anatomopatólogo especializado en sarcoma y grabados en una base de datos prospectiva en curso. Las características anatomopatológicas que fueron usadas para definir el grado incluyeron: índice mitótico, necrosis, celularidad, pleomorfismo y tipo y subtipo histológicos o diferenciación.
Muestras congeladas de tejido de los especímenes quirúrgicos fueron depositadas y se registró el tipo de tejido y la ubicación de la congelación en la base de datos, ligándoselos directamente con las variables del paciente, tumor, tratamiento y resultados. Los pacientes fueron seguidos por recidiva local, sistémica, sobrevida específica por enfermedad y sobrevida global. Se inició un sistema automatizado por el cual, si un paciente ingresado en la base de datos retornaba a la institución, se enviaba una nota de seguimiento automáticamente al equipo de manejo de datos de sarcoma. Los pacientes sin seguimiento en los 6 meses posteriores al último registro, fueron identificados mediante reportes dedicados, de manera que el seguimiento alejado pudiera obtenerse en el momento oportuno.
Cualquier factor asociado o predisponente, incluyendo enfermedades genéticas subyacentes, tales como neurofibromatosis, retinoblastoma hereditario o síndrome de Li-Fraumeni, o antecedentes de exposición a radiación, o la presencia de linfedema – todos ellos conocidos como causas asociadas – fueron cuidadosamente documentados. No obstante, estas bases de datos no son estáticas y nuevas entidades son descritas y la clasificación histológica evoluciona, a través de la identificación de subtipos que definen más precisamente la biología del tumor y los patrones de conducta. Avances recientes en la caracterización molecular, genética y citogénica del sarcoma de tejido blando, han mejorado y refinado el diagnóstico y la etiología.
Para esta presentación, los autores se han enfocado en la distribución y resultados por sitio corporal, tamaño y profundidad, influencia del grado e histología y utilización de variables combinadas de pronóstico, para la predicción de resultados. Se describen las consecuencias de la recidiva local y la influencia de la caracterización molecular sobre el diagnóstico y el manejo.
Resultados
Desde julio de 1982 hasta mayo de 2013, se ingresaron 10.000 pacientes en la base de datos prospectiva de sarcoma de tejido blando. Aproximadamente el 40% de las lesiones ocurrió en las extremidades, 38% en áreas visceral y retroperitoneal y el resto distribuido en todo el cuerpo (Tabla 1). En la distribución de las lesiones en las extremidades, el muslo fue el sitio dominante, presumiblemente basado en el volumen presente de tejido blando. La distribución por sexo, tamaño y grado, se muestra en la Tabla 1. El sitio primario no fue conocido con precisión en algunos pacientes que se presentaron para ser tratados quirúrgicamente por recidiva local o metastásica. Para el grado, los autores excluyeron los tumores de estroma gastrointestinal (GIST), que no son clasificados sino caracterizados por tamaño y tasa mitótica.
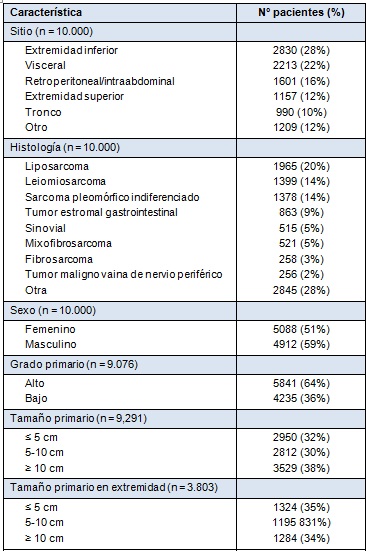
• TABLA 1: Distribución del 10.000 sarcomas de tejidos blandos en pacientes adultos admitidos para cirugía en el Sloan Kettering Cancer Center, entre el 1 de julio de 1982 y el 31 de mayo de 2013
El sitio es el mayor determinante del resultado y del patrón de recidiva. Las lesiones retroperitoneales e intraabdominales tienen propensión para la recidiva local temprana, con aproximadamente un 50% recidivando dentro de los primeros 5 años, seguido por una progresión continua e implacable, que se extiende por más de 20 años. Las lesiones en las extremidades y las viscerales, tienen una tasa de recidiva local de aproximadamente un 20% a 25% a 10 años, pero, en todos los sitios, la recidiva local tardía (>10 años) continúa ocurriendo, enfatizando la necesidad de un seguimiento alejado prolongado, dado que la recidiva local puede ocurrir a más de 2 décadas del diagnóstico inicial. Esos datos dan una descripción importante, no sólo de los resultados, sino también de la biología y patrones de conducta. Si se examina la sobrevida específica de la enfermedad, esto es, la muerte por la enfermedad, entonces el patrón es bastante diferente entre la lesión visceral y de la extremidad. Las lesiones en las extremidades, con una tasa de recidiva local de aproximadamente el 25% a 10 años, tienen una prevalencia de muerte específica por la enfermedad, de aproximadamente el 40% a 10 años, enfatizando la importancia de la enfermedad sistémica como causa de muerte.
Las lesiones retroperitoneales e intraabdominales, cuya recidiva local se extiende implacablemente por más de 15 años y excede el 60%, tienen un patrón de sobrevida específica de la enfermedad que es similar al patrón de la recidiva local, aunque con una contribución algo más pequeña de la enfermedad mestastásica. Más dramáticamente, las lesiones viscerales con una recidiva local de menos del 20% a 10 años, tienen una sobrevida específica de la enfermedad de aproximadamente el 35% a 10 años, enfatizando que la mayoría de esas muertes ocurre por enfermedad sistémica. La base de datos ha brindado, en efecto, información sobre el comportamiento biológico y patrón de recidiva o resultado global, esto es, que la muerte por lesiones en las extremidades y viscerales es predominantemente sistémica, mientras que en las lesiones retroperitoneales e intraabdominales, la causa dominante de muerte específica por la enfermedad, es la progresión local, aunque a menudo con progresión local multifocal.
Dos de los predictores más importantes de resultado son la histología/subtipo histológico y el grado. Para algunos tipos de sarcoma, el tipo histológico define el grado, con el sarcoma sinovial, rabdomiosarcoma y sarcoma de Ewing clasificados todos como de alto grado. Para el liposarcoma, los subtipos histológicos definen el grado, con el liposarcoma bien diferenciado y mixoide considerados como de bajo grado, y los liposarcomas desdiferenciado, de células redondas y pleomórfico, clasificados como de alto grado [2,3]. La distribución entre alto y bajo grado (~2:1) se ve en la Tabla 1. El grado es un predictor poderoso del resultado. Si se examina sólo a aquellos pacientes que se presentan con una lesión primaria (esto es, tratado primariamente en la institución) en la extremidad, hubo una tasa de recidiva local de aproximadamente un 20% a 10 años, para las lesiones de bajo y alto grado, mientras que la sobrevida específica de la enfermedad está altamente influenciada por el grado del tumor primario. La recidiva sistémica es infrecuente en las lesiones de bajo grado (<10%) a 20 años, mientras que en las lesiones de alto grado, la muerte por la enfermedad se acerca al 40% a 10 años. Nuevamente, la recidiva sistémica tardía (>10 años) y la muerte por la enfermedad, pueden ocurrir tanto en las lesiones de alto como de bajo grado. De interés: muchas de las lesiones iniciales de bajo grado que recidivan sistémicamente, pueden hacerlo con características de alto grado.
El tamaño y la profundidad son variables conocidas que pueden influenciar el resultado. No son necesariamente independientes, dado que es improbable tener un gran tumor superficial, mientras que se puede esperar que la mayoría de los tumores profundos tengan, al menos, 5 cm en su máxima dimensión. La distribución simple del tamaño primario se muestra en la Tabla 1 para todos los grados y presentaciones (primario y recidivas local y sistémica). Los pacientes con lesiones en las extremidades con un tamaño mayor a 10 cm tienen una sobrevida específica por la enfermedad menor al 40% a 15 años y la recidiva tardía y la muerte son vistas una vez más. Esto tiene distinciones importantes para los sistemas de estadificación, en donde el tamaño clásicamente es dividido entre menor a 5 cm y mayor a 5 cm. La combinación de tamaño y profundidad para las lesiones primarias de alto grado en la extremidad, enfatiza el impacto poderoso relativamente independiente del tamaño en relación con la profundidad, una vez que la lesión alcanza los 5 cm. y enfatiza la relativa rareza de las lesiones superficiales grandes (>5 cm), donde constituyen sólo el 8% de todas las lesiones mayores a 5 cm.
La distribución de la histología se muestra en la Tabla 1. Existe una relación dependiente del sitio con la histopatología, lo que se refleja en la dependencia del sitio de la metástasis inicial o dominante, con el sitio de la lesión primaria. Un ejemplo excelente es la dominancia del pulmón para el sitio primario en la extremidad y la predilección del leiomiosarcoma uterino por el pulmón, en contraposición con el GIST, que primariamente lo hace en el hígado. Las implicaciones para la sobrevida en el seguimiento alejado son obvias.
La histología es una variable que, con la progresión del tiempo y definición, se vuelve más precisamente definida. Un buen ejemplo es el leiomiosarcoma visceral, muchos de los cuales fueron clasificados como GIST, después del descubrimiento a fines de la década de 1990 de la activación de mutaciones en KIT y PDGFRA, específicamente en GIST. Esto proporciona conciencia de que esas bases de datos, en donde los datos inherentes pueden cambiar, se tornarán más variables. Esas bases de datos deben ser consideradas como entidades vivas y adaptables.
El diagnóstico de sarcoma de tejido blando ha sido revolucionado por el reconocimiento de las firmas diagnósticas moleculares. Eso no sólo se ve en muchas de las familias de tumores de Ewing, sino que también es aplicable a muchos otros subtipos histológicos [1]. Un buen ejemplo es el sarcoma sinovial, caracterizado por la traslocacion cromosómica t(X;18)(p11.1;q11.2) en casi todos los casos. Previamente considerado como un tumor que afectaba a adolescentes y adultos jóvenes, está claro en la actualidad que ese tumor puede ocurrir a lo largo de toda la vida. Más importante: considerado en el pasado como un tumor que ocurría en las extremidades y alrededor de las articulaciones, es claro actualmente que no deriva de la sinovia, aunque el tipo específico de célula es desconocido, y el tumor ha sido identificado en múltiples sitios inusuales, tales como la cabeza y cuello (menos del 10%), pared torácica o abdominal (menos del 10%) o sitios intratorácicos. Los autores han identificado esas lesiones especialmente en sitios inusuales, como el corazón, próstata y diafragma.
Las metástasis en ganglios linfáticos en el sarcoma de tejido blando son raras [4]. Cuando ocurre en ausencia de otras metástasis, algunos pacientes pueden ser rescatados. No obstante, la combinación de la presencia de una metástasis ganglionar linfática y otras metástasis es un signo de mal pronóstico.
Discusión
El presente reporte resalta la ventaja de tener grandes bases de datos enfocadas en entidades tumorales que son raras. Esos conjuntos de datos pueden ser utilizados no sólo para la predicción de factores pronósticos [3,5,6], sino también para la identificación de variados y variables eventos pronósticos al momento de la presentación, que influencian el tipo de recidiva, esto es, sobrevida específica relacionada con la enfermedad, sobrevida libre de metástasis o sobrevida global. Más importante: brindan una descripción estable de resultados específicos en pacientes, en una serie de enfermedades raras, brindando una línea de base para el resultado y riesgo previstos.
Esa identificación de variables prospectivas puede ser utilizada después, en la estratificación apropiada en ensayos controlados y randomizados [6]. Los autores de este trabajo han tomado esos datos para describir nomogramas predictivos específicos para los resultados [7]. El desarrollo de esos nomogramas es absolutamente dependiente de la validación por otras bases de datos, nacional [8] e internacionalmente [9]. Las bases de datos están mejor construidas si el conjunto de datos es grande, si hay eventos positivos y negativos, por ejemplo, vivo o muerto, relativamente distribuidos de forma equitativa y el seguimiento alejado es prolongado. El sarcoma ha sido una excelente fuente para esos conjuntos de datos y los autores los han adaptado para predecir recidivas locales [10], predecir sobrevida después de la recidiva local [11], y aplicar a histopatologías únicas, como el liposarcoma {12], GIST [13], sarcoma sinovial [14] y tumor desmoide {15]. Esos conjuntos de datos son una rica herramienta, requerida para el desarrollo de otros modelos matemáticos, utilizando otras técnicas estadísticas tales como Bayesian Belief Networks [16].
Una observación importante es el valor de esas bases de datos para definir la sobrevida en el seguimiento alejado. Un conocimiento del sitio primario o del primer lugar de metástasis, el pulmón, como se ha mostrado para las lesiones en las extremidades y el hígado, en el GIST, puede enfocar – durante el seguimiento – a los estudios por imágenes a un único sitio, reservando las imágenes adicionales para aquellos con positividad en el sitio dominante.
Los autores han comentado y documentado previamente, las consecuencias de la recidiva local. Claramente, una recidiva local grande temprana se acompaña por un diagnóstico mucho más pobre, que una recidiva tardía pequeña, particularmente si la recidiva inicial es de alto grado y la tardía de bajo grado [17]. Sin embargo, como se ha ilustrado en este trabajo y en un ensayo prospectivo randomizado [5], la disminución de la recidiva local no se traslada a un beneficio en la sobrevida específica por la enfermedad, un principio importante.
El diagnóstico molecular ha sido un avance extraordinario, como lo han demostrado los autores, utilizando como ejemplo al sarcoma sinovial. El sarcoma sinovial, previamente considerado como una entidad que ocurría en adolescentes y adultos jóvenes y alrededor de las articulaciones, ha sido caracterizado ahora por una fusión genética recurrente SYT-SSX [18]. La identificación de esa fusión genética por el diagnostico molecular [19,20] ha resultado, como se ha demostrado aquí, en la identificación de esos tumores en pacientes a lo largo de toda la vida, aunque con una predilección por los adolescentes y adultos jóvenes. No sólo se ha definido mejor el espectro de la edad, sino que se han caracterizado sitios inusuales previamente no concebidos como asociados con la “sinovia”.
Eso, por sí mismo, enfatiza la ausencia de conocimiento del origen celular para el “sarcoma sinovial”, que claramente no proviene de la sinovia. Esa identificación de firmas moleculares en el sarcoma y en otros tumores raros, se ha vuelto un potencial para la terapia. Si se puede demostrar que un tumor expresa una mutación genética como un oncogén o como un gen supresor del tumor, tal como el p53, o una expresión exagerada de un gen particular, que actúa conduciendo a una mutación, es obvio el potencial para la terapia. Eso está bien descrito en la identificación de la mutación c-kit en el GIST. La identificación de esa mutación y la habilidad de captar un inhibidor terapéutico de la tirosina-quinasa, como el imatinib, transformó el manejo de esos tumores. Históricamente [21], la mayoría de esos pacientes progresaba a una muerte temprana. Con el advenimiento de una terapia selectivamente dirigida, no solo se prolongó la sobrevida en presencia de enfermedad avanzada o metastásica [22], sino que el agente pudo ser utilizado en ensayos randomizados, como una terapia adyuvante, después de la resección quirúrgica [23]. Con el mayor conocimiento, se ha vuelto claro que cada droga selectiva actúa presumiblemente en contra, o al menos preferencialmente, contra una mutación, pero no sobre otras, como se ha visto en la diferencia entre la respuesta al exón 9 mutado y al exón 11 mutado del gen c-kit.
Como también han mostrado los autores en este trabajo, las bases de datos pueden convertirse en herramientas remarcables para examinar indicadores pronósticos. No sólo pueden identificarse nomogramas específicos del paciente, sino que el conjunto de datos puede ser examinado por otras asociaciones, utilizando herramientas como Bayesian Networks [16].
En esa situación, aún en ausencia de datos completos sobre cada paciente, se puede buscar matemáticamente asociaciones que pudieron no haber sido anticipadas cuando se piensa puramente en el contexto del pronóstico. Como también han demostrado los autores, esos datos pueden brindar las bases para herramientas predictivas, que pueden ser ulteriormente examinadas por el impacto de la significación de los antígenos de superficie, objetivos moleculares o incluso terapia. Se puede sugerir que, si la calidad del resultado predictivo tiene muy poca variación y se interviene con un evento terapéutico, como que el resultado observado excede por mucho los límites de confianza del resultado predictivo, se puede argumentar a favor de un efecto terapéutico. Si eso pudiera demostrarse, entonces se tendría una herramienta para utilizar en tumores raros e inusuales, en donde los ensayos randomizados controlados son esencialmente imposibles. Contrariamente, eso requiere un conjunto grande de datos sobre tumores raros y un seguimiento por un período prolongado.
Nuevas terapias dirigidas son desesperadamente necesarias para los aproximadamente 6.000 pacientes que mueren cada año en los Estados Unidos por sarcoma [26]. Un desafío clave será identificar las alteraciones que conducen a la sarcomagénesis para cada subtipo de sarcoma, de manera de poder identificar objetivos específicos de subtipos para la terapia. Para lograr esa meta, el grupo de estos autores realizó un análisis integrativo de la secuencia del ADN, número de copias y expresión del ADNm en 207 muestras, abarcando 7 subtipos mayores de sarcoma, que fueron vinculados con los datos clínicos desde la base de datos prospectiva. Frecuentemente, los genes mutados incluyen TP53 (17% de liposarcomas pleomórficos), NF1 (10,5% de mixofibrosarcomas y 8% de sarcomas pleomórficos) y PIK3CA (18% de liposarcoma mixoide/células redondas).
Las mutaciones PIK3CA en el liposarcoma mixoide/células redondas, se asociaron con activación Akt y con malos resultados clínicos. CDK4 y YEATS4 se encontraron ampliados y sobre-expresados en el liposarcoma desdiferenciado y condujeron a la proliferación del liposarcoma, en vistas funcionales realizas en las líneas celulares del liposarcoma desdiferenciado. El análisis de micromatriz de muestras de liposarcoma para la expresión de genes, ha sido vinculado con la base de datos clínicos y usado para derivar un predictor multigenético de la sobrevida libre de recidiva a distancia, e identificar los genes que conducen a la progresión del liposarcoma [28]. Ese conjunto de datos de expresión de genes demostró que el liposarcoma bien diferenciado y el desdiferenciado tienen activación del ciclo celular y del control de vías, incluyendo la regulación por incremento de CDK4, MDM2, CDK1, CDC7, TOP2A, PRC1, PLK1 y las ciclinas B1, B2 y E2 [29,30]. Por lo tanto, esas vías pueden ser útiles como agentes terapéuticos. En efecto, el nutlin-3a, un antagonista selectivo del MDM2, induce la apoptosis e inhibe la proliferación de las líneas celulares del liposarcoma desdiferenciado, en concentraciones que no afectan a las células madre normales derivadas de los adipocitos [29].
Asimismo, el PD0332991, un inhibidor selectivo del CDK4/CDK6, inhibe la proliferación induciendo el cese del ciclo celular G1y la senescencia en las líneas celulares del liposarcoma bien diferenciado y xenoinjertos. [27].En un ensayo reciente de fase II del PD0332991 en pacientes con liposarcoma avanzado CDK4 amplificado, el 66% de los pacientes tuvo una progresión libre de enfermedad a las 12 semanas y un subconjunto tuvo una respuesta radiográfica y enfermedad estable prolongada [31]. Esos resultados brindan un uso racional de los antagonistas de MDM2 y de los inhibidores de CDK4 en pacientes con liposarcoma bien diferenciado y desdiferenciado y muestran el valor de los recursos tisulares y de líneas celulares, vinculados con factores clínicos y patológicos, para el descubrimiento de una nueva terapia apuntando a un subtipo específico.
Esa situación puede ser ampliada a análisis genéticos y de micromatriz más extensos, brindando entonces modelos matemáticos adicionales [32], buscando eventos funcionales significativos, como las alteraciones en el número de copias o mutaciones, que comiencen a disecar las vías de señalización activadas, por las que los tumores son llevados a crecer.
Con ese análisis, se puede comenzar luego a ver los patrones de expresión que identifican tumores de lugares muy diferentes, por ejemplo, cabeza y cuello versus ovario, en los que no se hubiera concebido que estuvieran en alguna vía biológicamente similar. Ese abordaje es el paso siguiente, después de la transición desde un conocimiento de que el tratamiento de la enfermedad maligna se ha vuelto específico para la enfermedad y no para el órgano o disciplina. Actualmente, los autores pueden concebir diferentes lesiones en diversos sitios en el cuerpo, aún con apariencia histológica distinta, que revelan vías de progresión y crecimiento, que pueden ser inhibidas independientemente del sitio de origen del tumor.
Comentario y resumen objetivo: Dr. Rodolfo D. Altrudi