Reporte de Casos
Caso 1
Un adolescente inicialmente consultó a un ortopedista a los 4 años por dolor en pierna y dificultad para deambular. Se reveló displasia fibrosa del cráneo, costillas, brazos bilateral, fémur derecho y tibia. El paciente requirió de 2 procedimientos quirúrgicos descompresivos.
La evaluación endócrina reveló función tiroidea normal con lesión quística en ecografía típica de la observada en MAS. Tenía exceso de hormona de crecimiento por lo que necesitó tratamiento con un análogo de somatostatina.
La ecografía de los testículos identificó lesiones sólidas comúnmente encontrada en niños con MAS que representa hiperplasia de células de Leydig. El exámen cutáneo a los 11 años demostró un parche pequeño café con leche en parte lateral derecha de tórax (Fig 1A) y máculas hiperpigmentadas en labio inferior (Fig 1 B). Las máculas en el labio comenzaron a los 11 años, y desde entonces el paciente notó oscurecimiento de la pigmentación y nuevas máculas en los labios (Fig 1 C) y en mucosa bucal anterior bilateral.
Caso 2
Una mujer consulta por evaluación de pigmentación oral mucosa progresiva. Sana hasta los 18 años, cuando experimentó su primera fractura. Varios meses posteriores, la paciente se fracturó el fémur izquierdo al caminar. Posteriormente, ese año se diagnosticó MAS luego de una imagen radiológica que demostró displasia fibrosa poliostótica (PFD) involucrando el húmero bilateral, cráneo, maxilar izquierdo, y varias costillas.
La menarca fue a los 9 años. El ultrasonido de tiroides demostró cambios quísticos de la tiroides sin hipertiroidismo bioquímico.
El examen dermatológico reveló numerosas manchas café con leche en sien, abdomen derecho, espalda derecha, y parte posterior de muslo derecho, la mayoría de las cuáles presentes desde el nacimiento. Posteriormente desarrolló múltiples lentigines en pulpejos de primero y segundo dedos. La paciente notó máculas hiperpigmentadas en los labios (Fig 2 A) a los 18 años, que se oscurecieron en los últimos años. También notó nuevas máculas hiperpigmentadas en la gingiva, lengua y mucosa labial y bucal.
Caso 3.
Un adolescente se fracturó el metatarso izquierdo a los 3 años, y luego presentó una segunda fractura en el mismo pie ese año. Presentó máculas café con leche diagnosticándose MAS. Se reveló PFD que involucraba la base y parte anterior del cráneo, ambos húmeros, ambos cúbitos, fémures, pulgar izquierdo tibia izquierda y peroné, y metatarsos izquierdos.
El ultrasonido de testículo identificó lesión sólida y quística típica de MAS. El ultrasonido de tiroides reveló varios nódulos hipoecoicos, eutiroideo. Presentaba exceso de hormona de crecimiento por lo que recibió terapia con análogo de somatostatina. El exámen dermatológico reveló máculas café con leche de tamaño mediano en sien izquierda y sacro izquierdo y un parche más pequeño en parte inferior de abdomen derecho. Presentaba varias máculas hiperpigmentadas en mucosa bucal bilateral y labios que se manifestó a los 7 años de edad (Fig 2 B).
Caso 4
Una mujer con antecedentes de sangrado vaginal y maduración precoz mamaria en la niñez. Había sido evaluada a los 4 años cuando se le prescribió testolactone para retrazar la pubertad.
Se identificó displasia fibrosa poliostótica del cráneo. Durante la niñez desarrolló PFD en maxilar, paladar superior, alrededor del foramen óptico y en el canal auditivo derecho. Manifestó posteriormente pérdida de la visión debido a un quiste óseo aneurismático que comprimía el nervio óptico.
Se sometió a tiroidectomía por hipertiroidismo.
El exámen dermatológico a los 22 años reveló 3 máculas café con leche en parte posterior de cuello, muslo derecho y zona sacra. Esto ha estado presente desde el nacimiento. También presentaba máculas marrón oscuras en línea media de labio inferior (Fig 2 C) que las desarrolló a los 20 años y se oscurecieron progresivamente.
Desarrollo
El síndrome de Mc Cune Albright es causado por una mutación postcigótica en GNAS. Debido a que MAS es una enfermedad mosaico, el testeo molecular requiere de tejido afectado y no se realiza de rutina.
El diagnóstico se realiza con la presencia de al menos 2 de 3 hallazgos clínicos de pigmentación café con leche, endocrinopatías hiperfuncionantes y PFD. La pigmentación café con leche en MAS es debida al incremento de la producción de un segundo mensajero, AMP c que se activa por Gsf en los melanocitos soportando la mutación. Esto resulta en un incremento de la expresión de gen tirosinasa y de la producción de melanina.
Las lesiones pigmentadas son máculas marrones claras a oscuras que pueden ser segmentarias y tienden a respetar la línea media. Las localizaciones más comunes son parte posterior de cuello, sacro y cabeza. Aunque se han reportado en estudios previos las manchas café con leche ipsilateral a la displasia fibrosa en un estudio de 140 pacientes con MAS, la localización de la pigmentación cutánea no se asocia con el lado de compromiso óseo subyacente. El tamaño de la pigmentación café con leche no se correlaciona con la extensión de la enfermedad ósea. Estas lesiones pigmentarias pueden estar presentes desde el nacimiento o desarrollarse posteriormente, y no disminuyen con la edad.
La pigmentación de la mucosa oral se documentó en reportes de MAS temprano, pero MAS típicamente no se incluye en el diagnóstico diferencial de síndromes con lentigines orales (tabla).
Las lentigines mucosas adquiridas son hallazgos clínicos del síndrome de Laugier-Hunziker, pero el inicio de la pigmentación cutánea es típicamente en la adultez y no se asocia con enfermedades internas.
El inicio temprano de las lentigines mucosas es el hallazgo clásico de genodermatosis asociadas con disfunción endócrina, Carney Complex (CNC) y síndrome de Peutz-Jeghers (PJS).
El complejo de Carney es una enfermedad autosómica dominante asociada con una mutación que inactiva la proteína quinasa, AMPc dependiente, tipo 1, gen en cromosoma 17, como así también un segundo locus genético en el cromosoma 2p16 que produce un fenotipo menor. Entre los pacientes con CNC, el 20-40% desarrolla mixomas cardíacos, cutáneos y en mama.
Existe una fuerte asociación entre CNC y enfermedades endócrinas incluyendo síndrome de Cushing (30%), acromegalia (12%), prolactina elevada (64%), anormalidades tiroideas (75%), tumores de células de Sertoli (41%), cistoadenomas de ovario (14%) y lesiones pancreáticas ( 2.5%).
El síndrome de Peutz-Jeghers es una enfermedad autosómica dominante causada por inactivada mutación en el gen supresor tumoral serina/treonina quinasa 11 (STK 11). Desarrollan pólipos gastrointestinales (GI) y malignidad del tracto GI, como en otros sitios. Los pólipos GI son hamartomas benignos más comunes en el yeyuno, íleon, y estómago pero pueden ocasionar dolor abdominal periódico por intususcepción y obstrucción.
Tanto PJS y CNC se asocian con tumores de células de Sertoli que se pueden presentar como ginecomastia en varones como resultado de la actividad de la aromatasa de los tumores.
En CNC las lentigines están presentes en el 70-80% de los individuos afectados.
La distribución característica incluye labios, conjuntiva, mucosa genital. Las lentigines se desarrollan en la niñez, se oscurecen en la pubertad y disminuyen luego de la cuarta década de la vida. Los nevos azules pueden aparecer en el 40% de los pacientes, pero la pigmentación café con leche es un hallazgo menos preciso.
En PSP, las lentigines pueden presentarse en los dedos, palmas y plantas, mucosa oral, perianal y genital. Las lentigines orales en PJS se reportan en casi el 90% de los individuos y similar a CNC comienzan en la niñez y se incrementan en número y tamaño, y se oscurecen. Las descripciones tempranas de PJS reportan que las lentigines disminuyen luego de la tercera década, excepto las localizadas en la mucosa oral.
Los 4 pacientes con MAS en la presente serie desarrollaron pigmentación perioral entre los 7 y 20 años. Las manifestaciones clínicas compartidas en MAS, CNC y PJS sugieren vías comunes ocasionando pigmentación, enfermedad endócrina, y tumorogénesis.
Zacharin y col reportaron 4 pacientes con MAS, 2 con pigmentación perioral, y todos con pólipos GI que histológicamente simulaban los pólipos hamartomatosos observados en PJP. Los análisis genéticos revelaron mutaciones GNAS en 3 de 4 muestras gastrointestinales.
Collins y col reportaron también pólipos GI en 7 de 140 pacientes con MAS, sugiriendo un incremento de incidencia de hamartomas GI en MAS similar a los observados típicamente en PJS.
Recientemente, Gaujoux y col describieron lesiones hepatobiliares o pancreáticas en 6 de 19 pacientes con MAS, incluyendo 3 con neoplasias mucinosas papilares intraductales. El mismo grupo ha reportado una alta prevalencia de neoplasias mucinosas papilares intraductales en pacientes con CNC.
Se reportó una serie de 4 pacientes con pigmentación mucosa y hallazgos cutáneos, óseos y endócrinos clásicos. A diferencia de la pigmentación café con leche presente desde el nacimiento o durante la infancia, la pigmentación mucosa en MAS se desarrolla posteriormente (7-20 años ). La pigmentación oral puede sub reportarse por su manifestación tardía pero también se manifiesta en una minoría de pacientes con esta condición en mosaico. MAS debería incluirse en los diagnósticos diferenciales de síndromes asociados con lentigines en labios y mucosa oral, que incluye PJS, CNC, y la condición adquirida del síndrome Laugier-Hunziker.
Este hallazgo destaca la superposición clínica entre los síndromes de lentiginosis mucosa y en particular un riesgo común de desarrollar enfermedad endócrina y gastrointestinal en MAS, CNC y PJS.
Por último debido a esta condición mosaico con presentación variable que puede clínicamente retrasarse, el reconocimiento de la pigmentación oral como característica de MAS puede facilitar el diagnóstico temprano, que tiene el potencial de minimizar morbilidad asociada a MAS, incluyendo la displasia fibrosa, pubertad precoz, hipertiroidismo, hiperplasia de células de Leydig y Sertoli, exceso de hormona de crecimiento, pérdida renal de fosfatos mediada por FGF 23 y síndrome de Cushing.
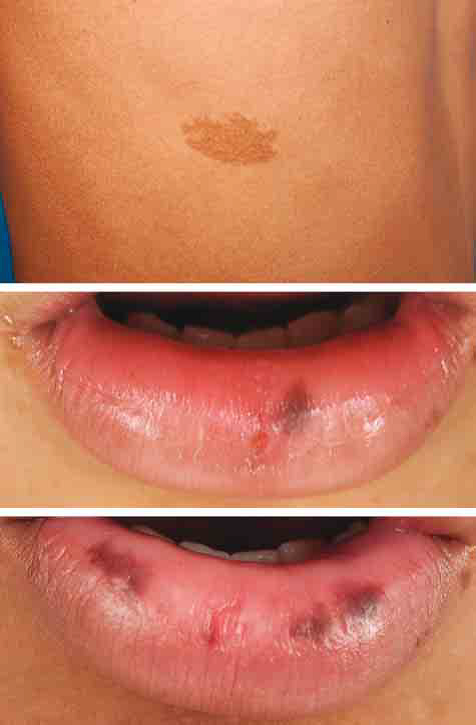
A, Parche café con leche en parte lateral derecha de tórax (edad 11 años). B, Pigmentación de la mucosa oral (edad, 11 años). C Pigmentación de la mucosa oral (edad 14 años).
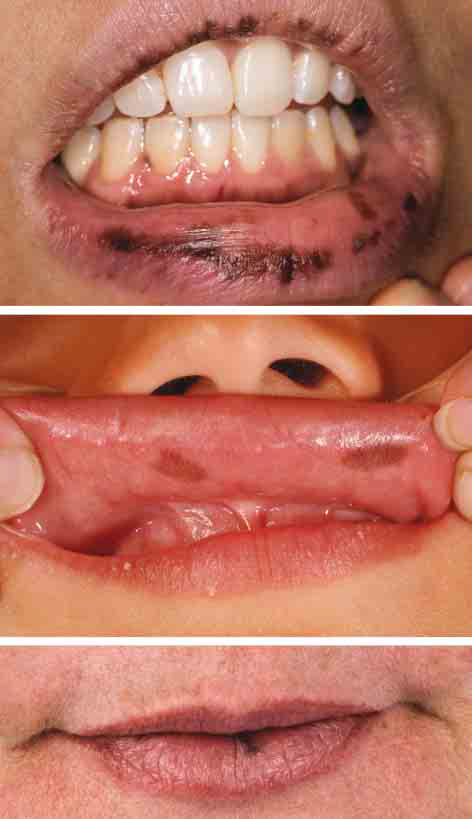
A, Caso 2. Pigmentación en bermellón y pigmentación mucosa (edad 26 años). B Caso 3 Pigmentación de la mucosa oral (edad 7 años). C Caso 4 Pigmentación de labio inferior (edad 28 años).
¿Qué aporta este artículo a la práctica dermatológica?
El síndrome de Mc Cune Albright (MAS) es una enfermedad esporádica caracterizada por displasis poliostótica fibrosa (PFD), endocrinopatías hiperfuncionantes y manchas café con leche en piel. Es causada por una mutación somática de la proteína de guanina nucleótido, del gen complex GNAS, ocasionando activación constitutiva de la subunidad alfa de la proteina G estimulatoria (Gsalfa). La pigmentación cutánea cubre generalmente una amplia área con un borde irregular, frecuentemente descripto como un borde de “la costa de Maine” y es generalmente el signo de presentación del síndrome.
(Comentario y resúmen objetivo: Dra. Geraldina Rodríguez Rivello)