Introducción
Las dermatosis purpúricas pigmentadas (PPDs) son un grupo de enfermedades crónicas de la piel caracterizadas por un infiltrado inflamatorio perivascular que afectan los capilares de la dermis papilar, y la presencia de eritrocitos extravasados con depósitos de hemosiderina.
Los subtipos de PPDs incluyen las dermatosis pigmentarias progresivas (enfermedad de Schamberg), púrpura anular telangiectásica (púrpura de Majocchi), dermatosis purpúrica pigmentada liquenoide de Gougerot y Blum, púrpura de Doucas y Kapetanakis, líquen aureus (líquen purpúrico).
Una de las menos caracterizadas es la PPD granulomatosa, primero reportada en 1996 por Saito y Matsuoka. Desde ese tiempo se reportaron 16 casos en la literatura. Originalmente se pensaba en una asociación entre hepatitis C, anticuerpos antinucleares o factor reumatoideo.
Actualmente parece haber una correlación positiva con hiperlipidemias y PPD granulomatosa, con 9 de 17 pacientes reportados (incluyendo el caso actual) presentando colesterol elevado.
Reporte
Se presenta una mujer de 59 años con antecedentes de rash pruriginoso de 3 semanas de evolución. El rash comenzó en las piernas y se extendió para comprometer tronco y brazos. Presentaba antecedentes de un presunto líquen plano oral (LP) que resolvió sin tratamiento. Recibió omeprazol por 6 años, pero lo cambió a dexlansoprazol 3 meses previos a la presentación. Recibía vitamina D, hace 5 meses.
Al exámen físico, presentaba numerosas pápulas eritematosas de 1-2 mm, distribuída difusamente en tronco y miembros (Fig 1a,b). Los diagnósticos diferenciales clínicos para las lesiones de las piernas incluían la enfermedad de Schamberg, mientras que las condiciones consideradas para brazos y tronco incluyeron el granuloma anular generalizado (GA), sarcoidosis papular, LP y erupción por drogas.
Se realizaron múltiples biopsias del brazo y espalda de la paciente. La evaluación histológia reveló cambios vacuolares e infiltrado de linfocitos. Rara disqueratosis sin epidermotropismo. En dermis superficial infiltrado nodular linfohistiocitario sin células multinucleadas, sugestivo de formación de granulomas (Fig 2).
Se observó extravasación de eritrocitos, con depósitos focales de hemosiderina. Los eosinófilos estaban ausentes.
- Los hallazgos de laboratorio incluyen elevaciones del colesterol total (273 mg/dL, normal menos de 200 mg/dL) y de LDL (175 mg/dL; menos de 100 mg/dL).
- Los tests de HDL, triglicéridos, glucemia y transaminasas fueron normales.
- Los tests para virus de hepatitis A, C y Ag de superficie para hepatitis B eran negativos.
- Aunque se consideraron etiologías infecciosas, las tinciones especiales y cultivos no encontraron hongos, bacterias ni micobacterias.
Las características fueron consistentes con la variante granulomatosa de PPD. Se le indicó tratamiento tópico con acetónido de fluocinolona. Al mes 10 de seguimiento las lesiones de las piernas mejoraron notablemente, y las de brazos y tronco ligeramente.
La variante granulomatosa de PPD fue primero reportada en 1996 por Saito y Matsuoa. Desde entonces se han reportado 16 casos en la literatura (tabla 1). Las lesiones son crónicas y se observan en piernas, siendo el dorso de pies el sitio más comúnmente afectado.
Sin embargo, se ha reportado el compromiso en otros sitios. Esta paciente presentaba PPD granulomatosa en espalda y es el tercer caso de PPD granulomatosa que tiene compromiso en otras áreas fuera de las extremidades.
Nueve de 17 pacientes (incluyendo el actual) con PPD granulomatosa presentaban colesterol elevado. En ateroesclerosis, la hiperlipidemias con depósitos de colesterol causa inflamaión vascular, que es el mecanismo propuesto en la patogénesis de la PPD granulomatosa.
La hiperlipidemias es común en la población de US, por lo que se necesitan de estudios futuros para determinar si esta asociación juega un rol en la patogénesis.
Las diferentes variantes clínicas de PPD demuestran cambios epidérmicos y tipos de infiltrados inflamatorios variables afectando los vasos de la dermis papilar.
Probablemente, la inflamación perivascular ocasione daño vascular y extravasación eritrocitaria.
En la PPD granulomatosa se observa un infiltrado linfohistiocitario con formación de granulomas en dermis papilar.
La presencia de granulomas formados es el hallazgo característico de PPD granulomatosa, cinco casos, incluyendo el presente caso, presentaban infiltrado linfohistiocitario en banda en la unión dermo epidérmica.
Varias condiciones deben considerarse en los diagnósticos diferenciales histológicos. Se deben excluir micobacterias típicas y atípicas, infección por hongos, aunque en casos infecciosos, el infiltrado linfohistiocitario se extiende más profundamente y los granulomas muestran caseosis central. La sarcoidosis muestra granulomas sin infiltrado liquenoide.
La erupción por drogas puede mostrar infiltrado linfohistiocitario con zonas con granulomas; la presencia de eosinófilos y la correlación clínico patológica con los antecedentes facilita el diagnóstico.
Puede haber una superposición histológica entre PPD y linfoma cutáneo de células T en parche (CTCL), como lo demostraron reportes de casos de estadío temprano de CTCL presentándose como PPD granulomatosa. Las claves diagnósticas a favor de CTCL incluye la presencia de grandes grupos de linfocitos en el estrato espinoso superior, linfocitos intraepidérmicos y linfocitos atípicos.
Los hallazgos clínicos pueden ser útiles, la PPD se localiza más frecuentemente en piernas. La monoclonalidad de células T se ha documentado en un subgrupo de pacientes con PPD sin compromiso de piernas clínicamente y con un infiltrado liquenoide histológicamente.
Además, algunos de estos pacientes es más probable que tengan concomitante o subsecuentemente desarrollen CTCT. Por lo tanto, los pacientes con PPDs requieren de un seguimiento a largo plazo clínico e histológico para monitorear la progresión a CTCL.
La PPD granulomatosa es similar a otras PPDs en la apariencia clínica pero se distingue histológicamente por un infiltrado linfohistiocitario con formación de granulomas.
Se presenta frecuentemente en piernas como máculas o pápulas rojo amarronadas. Debe excluirse otras causas de formación de granulomas como infección por hongos, micobacterias, sarcoidosis y granuloma anular.
Algunas veces se observa la presencia de infiltrado liquenoide en PPD granulomatosa, pero eleva sospecha de micosis fungoide. Parece haber una asociación entre PPD granulomatosa e hiperlipidemia.
Tabla 1 Resúmen de los casos reportados de dermatitis purpúrica pigmentada granulomatosa (PPD).

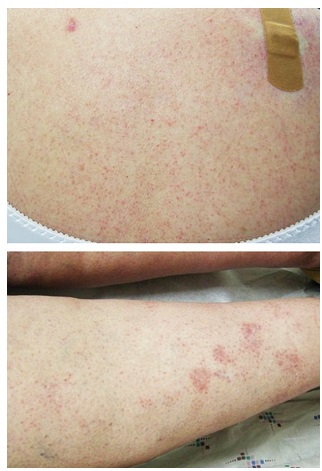
Figura 1 (a) Pápulas eritematosas distribuídas difusamente en parte superior de espalda;
(b) Pápulas eritematosas algunas formando placas en miembros inferiores.
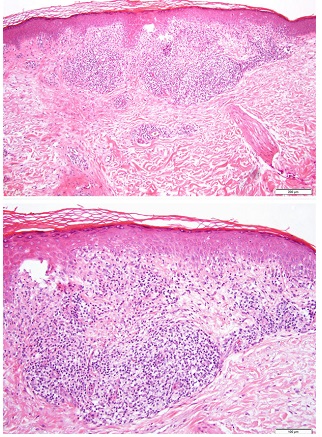
Figura 2 (a) Infiltrado linfohisticiotario superficial en dermis. Granulomas presentes; (b) colecciones de histiocitos formando granulomas, con extravasación de eritrocitos y depósitos focales de hemosiderina.
¿Qué aporta este artículo a la práctica dermatológica?
La variante granulomatosa de las dermatosis purpúricas pigmentadas (PPDs) es una condición rara e infrecuentemente descripta, con un total de 16 casos publicados en la actualidad. Se deben excluir otras enfermedades como infección atípica, sarcoidosis papular y granuloma anular generalizado.
Comentario y resúmen objetivo: Dra. Geraldina Rodríguez Rivello