| Introducción |
La enfermedad pulmonar intersticial (EPI) se caracteriza por inflamación y/o fibrosis en el intersticio alveolar del pulmón. Aproximadamente entre el 30% y el 40% de las personas con EPI desarrollan fibrosis pulmonar progresiva, que generalmente causa insuficiencia respiratoria y se asocia con una supervivencia media de aproximadamente 2,5 a 3,5 años. Esta revisión resume la evidencia actual sobre el diagnóstico y el tratamiento de la EPI.
| Métodos |
Se realizó una búsqueda en PubMed de estudios en idioma inglés sobre la epidemiología, fisiopatología, diagnóstico, tratamiento y pronóstico de la enfermedad pulmonar intersticial publicados entre el 1 de enero de 2010 y el 15 de enero de 2024. Se inspeccionaron manualmente las listas de referencias de los artículos seleccionados en busca de otros artículos relevantes. Se recuperaron un total de 10 728 artículos. De los 728 artículos identificados, se incluyeron 115, que consistían en 38 ensayos clínicos, 7 artículos de revisión, 7 metanálisis, 22 estudios observacionales longitudinales, 34 estudios transversales y 7 guías, declaraciones científicas o documentos de consenso.
| Clasificación y nomenclatura en las EPI |
Las EPI se subcategorizan según la etiología e incluyen la EPI asociada a la enfermedad del tejido conectivo (ETC-EPI), la neumonitis por hipersensibilidad, la EPI inducida por fármacos, la EPI posinfecciosa y las neumonías intersticiales idiopáticas. Estas afecciones, que tienen características clínicas similares, se definen por apariencias histopatológicas y pronósticos distintos (Figura 1). Las EPI más comunes son la fibrosis pulmonar idiopática (FPI) (que representa >30 % de las EPI), la neumonitis por hipersensibilidad (que representa aproximadamente el 15 % de las EPI) y la enfermedad del tejido conectivo (ETC) (que representa aproximadamente el 25 % de las EPI).
Otras EPI incluyen la EPI inducida por fármacos y la EPI posinfecciosa (p. ej., posterior a COVID-19). Aunque los diferentes tipos de EPI tienen una fisiopatología, manifestaciones clínicas y pronósticos distintos, todas las formas de EPI pueden causar fibrosis pulmonar irreversible. Una vez establecida, la fibrosis pulmonar puede progresar incluso cuando se haya tratado o eliminado la causa subyacente de la EPI. La frase fibrosis pulmonar progresiva (FPP) se refiere al comportamiento de la enfermedad en un subconjunto de personas con EPI que se puede definir y tratar con mayor precisión en función del empeoramiento de la fibrosis en lugar de la etiología inicial. Las personas con EPI-ETC que cumplen los criterios de FPP tienen una supervivencia media de aproximadamente 4 años en comparación con una supervivencia media de 8 a 10 años para todos los pacientes con EPI-ETC.
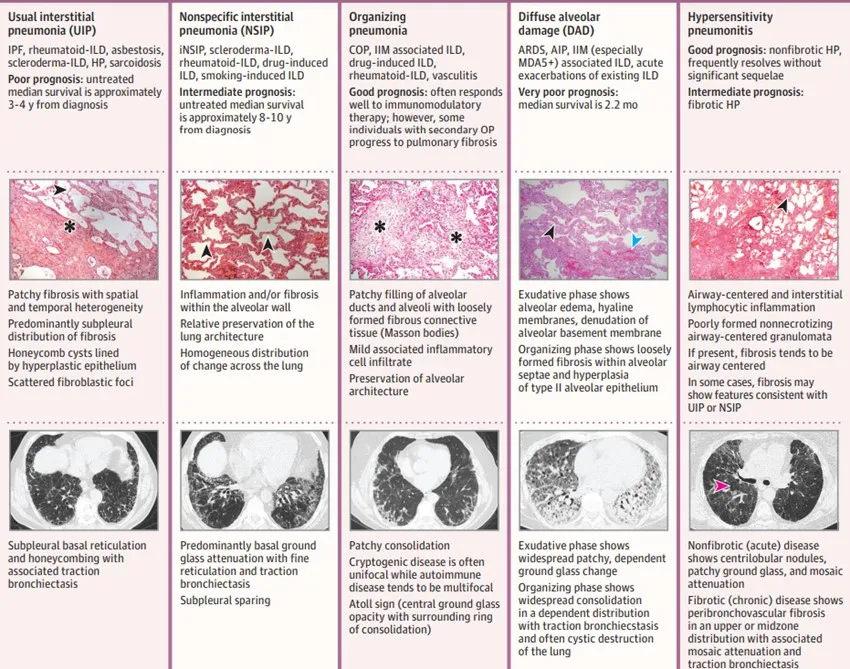
Figura 1. Características de los patrones histológicos de EPI más comunes
| Epidemiología |
La prevalencia de la EPI aumenta con la edad y es más alta en personas de entre 80 y 84 años. La edad media en el momento del diagnóstico es de aproximadamente 67 a 72 años. La incidencia de la FPI en adultos es de aproximadamente 3 a 9 por 100 000 y, en contraste con la EPI en general, es más común en hombres que en mujeres, con una razón de prevalencia de 3:1. Los estudios de casos y controles informaron que la exposición a polvos de madera, polvos metálicos, tabaquismo y contaminación urbana se asociaron con un mayor riesgo de FPI. La neumonitis por hipersensibilidad es una forma de EPI que se desencadena por la inhalación de antígenos específicos, más comúnmente proteínas aviares y esporas de hongos o moho. A diferencia de la FPI, la prevalencia de la neumonitis por hipersensibilidad es aproximadamente igual en hombres y mujeres.
La prevalencia de la EPI-ETC varía según la enfermedad subyacente. Aproximadamente el 65 % de todos los pacientes con esclerosis sistémica y el 80 % de los individuos con esclerosis sistémica cutánea difusa desarrollan EPI. Se estima que entre el 36 % y el 45 % de los individuos con miopatía inflamatoria idiopática (un grupo de trastornos que incluye polimiositis, dermatomiositis y síndrome antisintetasa) desarrollan EPI, pero la incidencia es tan alta como el 80 % en individuos con anticuerpos antisintetasa específicos. Aproximadamente entre el 52% y el 67% de los pacientes con enfermedad mixta del tejido conectivo, entre el 11% y el 27% de las personas con síndrome de Sjögren, entre el 1,5% y el 5% de las personas con artritis reumatoide y aproximadamente entre el 1% y el 2% de los pacientes con LES desarrollan EPI.
El índice de género, edad y fisiología pulmonar (GAP) integra información de la CVF y la capacidad de difusión de los pulmones para el monóxido de carbono (DLCO) (junto con la edad y el sexo) en pacientes con EPI fibrótica para estimar la supervivencia en función de 3 estadios de gravedad. El estadio 1 de GAP, que representa aproximadamente el 50% de los pacientes, se asocia con una mortalidad a 1 año del 5,6% y una mortalidad a 3 años del 16,3%. Aproximadamente el 10% de los pacientes recién diagnosticados tienen estadio 3 de GAP, que se asocia con una mortalidad a 1 año del 39,2% y una mortalidad a 3 años del 76,8%.
La fibrosis pulmonar idiopática (FPI) y otras EPI asociadas con fibrosis pulmonar se asocian con complicaciones relacionadas con la enfermedad, como hipertensión pulmonar y cáncer de pulmón. Aproximadamente un tercio de los pacientes con diagnóstico reciente de EPI fibrótica presentan evidencia de apnea obstructiva del sueño en la polisomnografía. Aproximadamente el 14 % de las personas con diagnóstico reciente de FPI tienen hipertensión pulmonar, y esta cifra aumenta a aproximadamente el 86 % en las personas con fibrosis pulmonar que esperan un trasplante de pulmón.
La incidencia de cáncer de pulmón en personas con EPI es de aproximadamente 25,2 casos por 1000 personas-año, una tasa al menos 3 veces mayor que la de las personas sin EPI de la misma edad y antecedentes de tabaquismo. Las personas con fibrosis pulmonar son susceptibles a exacerbaciones agudas, caracterizadas por un rápido empeoramiento de la disnea durante varios días o semanas. En estos pacientes, la tomografía computarizada (TC) de los pulmones muestra un cambio generalizado en vidrio esmerilado, que refleja el desarrollo de una lesión pulmonar aguda y daño alveolar difuso (Figura 1). En pacientes con FPI, la incidencia de exacerbaciones agudas a 1 año es de aproximadamente el 14,2% y la incidencia de exacerbaciones agudas a 3 años es de aproximadamente el 20,7%. Las exacerbaciones agudas de la FPI se asocian con resultados muy malos con una supervivencia media estimada de 2,2 meses.
| Fisiopatología |
La FPI se desarrolla debido a una respuesta anormal de cicatrización en individuos genéticamente susceptibles después de una lesión epitelial alveolar repetida. Aproximadamente entre el 2% y el 5% de los individuos con EPI fibrótica tienen otros familiares con EPI. La neumonitis por hipersensibilidad se caracteriza por una respuesta granulomatosa inmunomediada al antígeno inhalado.
En la esclerodermia y la miopatía inflamatoria idiopática, los autoanticuerpos específicos se asocian con el desarrollo de la EPI. En la artritis reumatoide, el perfil genético de los individuos que desarrollan EPI es similar al asociado con la FPI. Los fármacos y las infecciones también pueden precipitar la EPI. La bleomicina, la amiodarona, la nitrofurantoína y las inmunoterapias contra el cáncer, son los fármacos más comunes asociados con el desarrollo de la EPI.
Los infiltrados intersticiales asociados con la COVID-19 suelen resolverse sin tratamiento. Sin embargo, algunas personas con COVID-19 han desarrollado fibrosis pulmonar progresiva. Otros coronavirus, incluidos el MERS y el SARS, también se asociaron con el desarrollo de la EPI.
| Presentación clínica |
La disnea es el síntoma principal de la EPI, que generalmente se manifiesta primero durante un esfuerzo extenuante. A medida que la EPI progresa, las personas a menudo informan un deterioro gradual de la capacidad de ejercicio. En la enfermedad avanzada, la hipoxemia en reposo es común. Aproximadamente del 30% al 50% de los pacientes con FPI informan una tos que afecta la calidad de vida. A medida que la enfermedad pulmonar intersticial progresa, los pacientes suelen referir fatiga y pérdida de peso. La pérdida de peso no intencionada en pacientes con enfermedad pulmonar intersticial se asocia con un mal pronóstico. Una disminución de peso superior al 5 % en cualquier momento durante el curso de la enfermedad se asocia con un aumento de 2,5 veces en el riesgo de mortalidad.
La mayoría de los pacientes con enfermedad pulmonar intersticial debida a EPOC son diagnosticados con ésta antes de desarrollar síntomas respiratorios relacionados con la enfermedad pulmonar intersticial. Sin embargo, la enfermedad pulmonar intersticial puede ser la primera manifestación de una enfermedad autoinmune sistémica en una pequeña proporción de individuos. Por lo tanto, los médicos deben preguntar acerca de los síntomas sistémicos en individuos que presentan enfermedad pulmonar intersticial. La anamnesis debe evaluar la exposición a posibles factores desencadenantes de neumonitis por hipersensibilidad o exposiciones ocupacionales asociadas con el desarrollo de neumoconiosis como la silicosis o la asbestosis.
| Evaluación y diagnóstico |
En la presentación, aproximadamente entre el 7% y el 42% de los individuos con fibrosis pulmonar tienen hipocratismo digital. En la auscultación torácica, de los pacientes con FPI y el 73% de aquellos con EPI sin FPI tienen crepitantes finos tipo velcro en las bases pulmonares. Aquellos con neumonitis por hipersensibilidad pueden tener graznidos agudos al final de la inspiración en la auscultación pulmonar. Signos de ETC como artritis activa, engrosamiento de la piel y pápulas de Gottron pueden estar presentes en el examen físico.
Los pacientes con EPI fibrótica terminal pueden tener evidencia de cianosis o hallazgos clínicos de hipertensión pulmonar como un segundo ruido cardíaco (R2) fuerte, R3 o R4 (un ritmo de galope), presión venosa yugular elevada y edema periférico. La sensibilidad de la radiografía de tórax para la EPI es del 63% y la especificidad es del 93%. Las pruebas serológicas para autoanticuerpos y anticuerpos IgG específicos del suero (precipitinas) pueden sugerir enfermedad del tejido conectivo o neumonitis por hipersensibilidad como diagnósticos potenciales.
La TC torácica es la prueba diagnóstica primaria para identificar y diagnosticar la EPI con aproximadamente un 91% de sensibilidad y un 71% de especificidad para los subtipos de EPI. Sin embargo, ni la TC ni la histopatología por sí solas son diagnósticas para EPI específicas. Existe una superposición entre los diferentes hallazgos histopatológicos y radiológicos. El enfoque aceptado para el diagnóstico de EPI es la evaluación multidisciplinaria con un equipo formado por neumólogos, radiólogos y, cuando sea necesario, patólogos y reumatólogos.
Si bien la biopsia pulmonar era importante anteriormente para el diagnóstico preciso de EPI, más recientemente, la TC torácica ha reemplazado a la biopsia pulmonar, principalmente debido a las mejoras en las imágenes de TC. La biopsia pulmonar, en particular la biopsia pulmonar quirúrgica, se asocia con una tasa de mortalidad del 1% al 2%.
La criobiopsia transbronquial broncoscópica, un procedimiento endoscópico mínimamente invasivo, ha reemplazado a la biopsia torácica quirúrgica para obtener muestras de tejido pulmonar, con una tasa de complicaciones menor que la cirugía y un nivel similar de precisión diagnóstica. En una revisión sistemática de estudios publicados, la criobiopsia se asoció con efectos adversos de sangrado en el 30% de los pacientes y neumotórax en el 8% de los pacientes. Los efectos adversos graves, incluida la mortalidad, fueron poco frecuentes.
Incluso después de una evaluación clínica detallada que incluye una biopsia pulmonar, hasta el 15% de las personas tienen EPI que no se puede clasificar. En algunos pacientes con EPI inclasificable, el diagnóstico se vuelve más claro cuando aparecen nuevos signos y síntomas clínicos con el tiempo. Cuando se sospecha o se realiza un diagnóstico de EPI, se deben obtener pruebas de función pulmonar para evaluar la gravedad de la enfermedad. Los pacientes con EPI suelen presentar un patrón restrictivo en la espirometría. Una pérdida del 5% o más de la CVF durante 3, 6 o 12 meses se asocia con un peor pronóstico en comparación con una pérdida de la CVF inferior al 5 %, en pacientes con FPI, EPI asociada a esclerosis sistémica y todas las demás EPI fibróticas.
| Tratamiento |
> Terapia antifibrótica
La pirfenidona es un derivado de piridina que se administra por vía oral y tiene propiedades antiinflamatorias, antioxidantes y antifibróticas. En el ensayo clínico ASCEND, la pirfenidona redujo la disminución de la CVF en el seguimiento de 52 semanas. Los efectos adversos más comunes asociados con la pirfenidona en comparación con el placebo fueron erupción cutánea fotosensible, náuseas y anorexia. En un análisis agrupado que incluyó a 1247 pacientes, 22 pacientes que recibieron pirfenidona habían muerto en la semana 72 en comparación con 42 pacientes que recibieron placebo. En el ensayo clínico RELIEF de 127 pacientes con fibrosis pulmonar progresiva, la pirfenidona desaceleró la disminución de la CVF en comparación con placebo en un 3,53%
Nintedanib es un inhibidor oral de la tirosina quinasa de moléculas pequeñas. En un análisis de datos combinados de 2 ensayos clínicos aleatorizados (ECA) paralelos, INPULSIS-1 e INPULSIS-2, 1066 pacientes fueron aleatorizados a nintedanib o placebo. En el seguimiento de 52 semanas, la disminución de la CVF fue significativamente menor en el grupo de nintedanib. En INPULSIS-1, se produjo diarrea en el 61,5 % de los pacientes asignados aleatoriamente a nintedanib y en el 18,6 % de los pacientes asignados aleatoriamente a placebo. Las tasas correspondientes de diarrea en INPULSIS-2 fueron del 63,2 % en los pacientes asignados aleatoriamente a nintedanib y del 18,3 % en los pacientes asignados aleatoriamente a placebo. En el ensayo clínico aleatorizado SENSCIS de 579 pacientes con enfermedad pulmonar intersticial asociada a esclerosis sistémica, nintedanib redujo la disminución de la CVF en comparación con placebo. En un análisis post hoc, los pacientes con la mayor reducción en la disminución anual de la CVF recibieron la combinación de nintedanib y micofenolato de mofetilo. En el ensayo clínico aleatorizado INBUILD de 663 pacientes con FPP debido a una causa distinta a la FPI, nintedanib redujo significativamente la disminución de la CVF en comparación con placebo.
> Terapia inmunomoduladora
En los ensayos clínicos aleatorizados FaSScinate y FocuSSced de individuos con esclerosis sistémica cutánea difusa e inflamación activa (definida como artritis, plaquetas elevadas o proteína C reactiva elevada), tocilizumab no mejoró significativamente el resultado primario del cambio a las 48 semanas en la escala de Rodnan modificada. Ninguno de los estudios requirió la presencia de EPI. En ambos estudios, el punto final secundario del cambio a las 48 semanas en la CVF sugirió un beneficio de la terapia que fue suficiente para que la FDA lo aprobara como tratamiento para la EPI-esclerodermia en los EE. UU.
El ensayo Scleroderma Lung Study 1, la ciclofosfamida mejoró significativamente el % previsto de la CVF en comparación con placebo. El ensayo Scleroderma Lung Study 2 comparó micofenolato de mofetilo oral 1,5 g dos veces al día con ciclofosfamida oral en individuos con EPI asociada a esclerosis sistémica y no encontró diferencias en el % previsto de la CVF entre los grupos; menos pacientes del grupo de micofenolato de mofetilo interrumpieron el tratamiento a los 12 meses (29% frente a 43,8%). En el ECA multicéntrico DESIRES, el rituximab redujo significativamente la puntuación cutánea de Rodnan modificada (resultado principal) en comparación con placebo en 6,3 en comparación con 2,14. Entre los participantes con una CVF inferior al 80 % al inicio, el rituximab mejoró el % de CVF previsto a las 24 semanas en comparación con placebo.
En el ensayo clínico aleatorizado, doble ciego y doble simulación RECITAL, se comparó el rituximab con la ciclofosfamida en 101 personas con enfermedad pulmonar intersticial (EPI) debida a esclerosis sistémica, enfermedad mixta del tejido conectivo o miositis inflamatoria idiopática. El rituximab no fue significativamente mejor que la ciclofosfamida en cuanto al resultado primario del cambio en la CVF a las 24 semanas. Sin embargo, ambos fármacos aumentaron la CVF y mejoraron la calidad de vida.
Aunque los corticosteroides y las terapias inmunosupresoras como la azatioprina y el micofenolato de mofetilo se prescriben con frecuencia para tratar la neumonitis por hipersensibilidad y la artritis reumatoide (EPI), no se han probado en ECAs en ninguna de las dos enfermedades. Aunque faltan farmacoterapias basadas en evidencia para la neumonitis por hipersensibilidad, evitar una causa desencadenante identificada (como las aves o el moho) puede mejorar la afección. En el caso de la artritis reumatoide (EPI), los datos observacionales sugirieron que el rituximab, el abatacept y el tofacitinib se asociaron con los mejores resultados pulmonares, incluida una menor incidencia de EPI y menos hospitalizaciones respiratorias.
> Trasplante
El trasplante de pulmón es una opción terapéutica para personas con enfermedades pulmonares en etapa terminal, incluida la EPI. La edad avanzada y las comorbilidades como la enfermedad cardiovascular, la diabetes y la disfunción cardíaca derecha excluyen el trasplante como una opción terapéutica en muchos pacientes con EPI. En el informe de 2019 de la International Society for Heart and Lung Transplantation, la supervivencia media fue de 6,2 años para todos los receptores de trasplante de pulmón.
| Hipertensión pulmonar |
Pocos tratamientos mejoran los resultados en pacientes con EPI e hipertensión pulmonar. En el ensayo clínico aleatorio INCREASE de 326 pacientes con EPI e hipertensión pulmonar, el treprostinil aumentó la distancia recorrida en la prueba de caminata de 6 minutos a las 16 semanas en comparación con placebo. Los eventos adversos más comunes fueron tos transitoria (43,6 %), cefalea (27,6 %), irritación de garganta (12,3 %) y dolor orofaríngeo (11,0 %).
| Rehabilitación pulmonar |
La rehabilitación pulmonar, que consiste en un programa estructurado de educación y entrenamiento de resistencia durante 8 a 12 semanas, es una terapia eficaz para mejorar la capacidad de ejercicio y reducir los síntomas en personas con enfermedad pulmonar crónica. Un metaanálisis Cochrane de 2021 informó que en personas con disnea sintomática debido a EPI, la rehabilitación pulmonar se asoció con una mejora en la distancia de la prueba de caminata de 6 minutos en comparación con el control. La rehabilitación pulmonar se asoció con una mejora de los síntomas de disnea y una mejora de la calidad de vida relacionada con la salud.
| Terapia de apoyo y basada en los síntomas |
Los pacientes con enfermedad pulmonar crónica deben vacunarse contra el neumococo, la COVID-19, el virus respiratorio sincitial y la gripe. Las personas que fuman actualmente deben recibir ayuda para dejar de fumar. La oxigenoterapia ambulatoria y continua son tratamientos importantes para los pacientes con enfermedad pulmonar intersticial avanzada. En el ensayo prospectivo AMBOX, el oxígeno se asoció con una mejora clínicamente significativa en la calidad de vida medida con el K-BILD. La oxigenoterapia de 24 horas es adecuada para pacientes con enfermedad pulmonar intersticial que tienen saturaciones de oxígeno en reposo consistentemente inferiores al 90%.
La tos y la disnea son comunes en pacientes con EPI. Los opiáceos de acción corta mejoran la disnea en pacientes con enfermedad pulmonar terminal. La nalbufina, un agonista-antagonista opioide, redujo la frecuencia objetiva de la tos en comparación con placebo. La morfina redujo la frecuencia de la tos en un 39,4% en comparación con placebo en un estudio cruzado de 2 semanas. El efecto adverso más frecuente fue el estreñimiento (21%). Las benzodiazepinas pueden mejorar la disnea aguda y los episodios de pánico en personas con enfermedad terminal; sin embargo, la evidencia de los RCT es mixta con respecto al beneficio de esta clase de medicamentos.
La planificación del final de la vida y el acceso a servicios de cuidados paliativos son importantes para las personas con enfermedad pulmonar intersticial e insuficiencia respiratoria. Las directivas anticipadas brindan a los pacientes control sobre sus tratamientos y resultados a medida que progresa su enfermedad. Para las personas con enfermedad terminal para quienes el trasplante de pulmón no es una opción, se debe evitar la intubación y la ventilación mecánica porque están asociadas con malos resultados.
| Pronóstico |
La supervivencia media de las personas con FPI es de 3 a 3,5 años. El análisis de los ECA de pirfenidona y nintedanib, realizado por la FDA, encontró que una mayor disminución de la CVF se asoció con un mayor riesgo de mortalidad. La desaceleración de la disminución de la CVF con pirfenidona o nintedanib se asoció con una mejora de la expectativa de vida de aproximadamente 1 a 2,5 años.
| Conclusiones |
La enfermedad pulmonar intersticial generalmente se presenta con disnea de esfuerzo y puede progresar a insuficiencia respiratoria. El tratamiento de primera línea es nintedanib o pirfenidona para la fibrosis pulmonar idiopática (FPI) y micofenolato de mofetilo para la enfermedad pulmonar intersticial (EPI) debida a enfermedad del tejido conectivo. Se debe considerar el trasplante de pulmón para pacientes con EPI avanzada. En pacientes con EPI, el entrenamiento físico mejora la distancia recorrida en la prueba de caminata de 6 minutos y la calidad de vida.
Traducción y resumen objetivo: Dr. Cristian Pisa