|
Resumen La síntesis de novo de colesterol y su enzima limitante, hidroximetilglutaril-coenzima A reductasa (HMGCR), es crítica para la regulación de la proliferación y la supervivencia celular en cáncer. El objetivo general del presente trabajo es estudiar el papel de la vía del colesterol en la adquisición de propiedades de células madre en modelos de cáncer de mama (CM). Con ese propósito, se desarrolló un modelo de sobreexpresión de HMGCR en la línea celular MCF-7 utilizando un sistema CRISPR activador (CRISPRon). La generación de dicho sistema comprendió las siguientes etapas: 1. Diseño y síntesis de las sondas (sgRNAs) 2. Clonación de las sondas 3. Transformación de bacterias con los plásmidos clonados 4. Verificación de dichas construcciones por secuenciación 5. Transfección de líneas celulares 6. Caracterización del sistema. La expresión de HMGCR aumentó significativamente en las células MCF-7/CR en comparación al control de transfección (MCF-7/TC), tanto a nivel transcripcional como proteico, y se observan niveles similares a los detectados en otros modelos de células madre (normales y tumorales). El fenotipo HMGCRon se asoció a un incremento de los niveles de los marcadores de pluripotencia Nanog y Sox2, un aumento en la población CD44+/CD24- y CD133+ y un aumento en la frecuencia de formación de mamoesferas, características asociadas a células madre tumorales (CMTs) en CM. A continuación, se evaluó la viabilidad de estas células en respuesta al tratamiento con simvastatina (SIM) y lovastatina (Lova). Se encontró que las células MCF-7/CR muestran mayor sensibilidad a SIM que la línea control MCF-7/TC. Estos datos sugieren que la expresión de HMGCR está asociada a características de CMTs y determina, al menos en parte, la sensibilidad a estatinas, sentando las bases para estudios posteriores orientados a una terapéutica anti-colesterol específica para el compartimiento de células madre en CM. |
| Introducción |
> Células Madre Tumorales
Según el modelo de Células Madre Tumorales (CMTs), el crecimiento del tumor, al igual que el desarrollo normal de cualquier tejido, depende exclusivamente de pocas células que componen el compartimiento de células madre. Por lo tanto, la mayoría de las células del tumor no poseen la capacidad de autorrenovación y por consiguiente no contribuyen a la perpetuación del mismo. En este modelo, la heterogeneidad tumoral surge exclusivamente de la diferenciación aberrante de las CMTs(1)(2).
Se considera que las CMTs tienen las mismas propiedades que las células madre normales: autorrenovación, resistencia a químicos, pueden permanecer en quiescencia por períodos largos de tiempo y son capaces de colonizar otras partes del cuerpo(3). Por lo tanto, este modelo parece explicar la resistencia a las terapias convencionales, recurrencia del tumor e incluso la metástasis(4). Sin embargo, este concepto “clásico o estático” conlleva la presunción de que la población de CMTs se mantiene estable e invariable a lo largo del tiempo y que el “estado de CMT” no puede ser adquirido por una célula tumoral diferenciada. Por el contrario, estudios recientes sugieren que el fenotipo de las CMTs es fluido y puede ser regulado(4).
En el desarrollo normal, la plasticidad fenotípica de las células se encuentra altamente regulada. Por otro lado, las células tumorales diferenciadas son capaces de reactivar esta propiedad y adquirir un fenotipo de CMT. Las CMTs pueden cambiar de un estado CMT a uno no-CMT de manera reversible, dependiente de estímulos endógenos y exógenos, como por ejemplo cambios en el metabolismo celular(5). A este modelo se lo conoce como “Modelo dinámico de CMTs”.
Al-Hajj y colaboradores demostraron por primera vez la existencia de las CMTs en CM, al identificar que tan sólo 100 células CD44+/CD24-/low eran capaces de formar tumores en ratones inmunosuprimidos, mientras que células con otros fenotipos eran incapaces de hacerlo. Los tumores generados en los ratones presentaban las mismas características del tumor del paciente del cual las células fueron aisladas, y además, podían ser pasados serialmente, demostrando su capacidad de autorrenovación(6).
A pesar de la falta de consenso en cuanto al fenotipo y frecuencia de las CMTs en la mayoría de los tumores sólidos, en los años siguientes a esta publicación, la evidencia apoyando la existencia de CMTs en este tipo de tumores fue ampliándose para incluir cerebro, colon, cuello y cabeza, páncreas, pulmón, melanoma y tumores mesenquimales y hepáticos(7)(8).
Las CMTs en mama pueden ser identificadas y aisladas por diferentes metodologías, incluyendo citometría de flujo para marcadores de superficie específicos, como CD44, CD24 y CD133, formación de mamoesferas en suspensión y actividad de la enzima aldehído deshidrogenasa (ensayo ALDEFLUOR)(9)(10)(11).
Además, algunos estudios han implicado a la transición epitelio-mesenquimal (EMT, Epithelial- Mesenchymal Transition) y a la transición mesenquimal-epitelial (MET, Mesenchymal- Epithelial Transition) en la generación de estados de CMTs. Estos trabajos identifican una subpoblación dentro del grupo de las CMTs que presenta características del tipo mesenquimal, potencial invasivo y se caracteriza por la expresión de los marcadores CD44+/ CD24-.
Por otro lado, existe una subpoblación proliferativa de CMTs con marcación positiva para ALDH y que se asemejan a un tipo más epitelial. Cabe destacar que, debido a la plasticidad fenotípica, estas células son capaces de hacer la transición entre estos estados de CMTs, en línea con el modelo dinámico(12).
Por otro lado, algunos de los factores utilizados en la generación de células madre con pluripotencia inducida (iPSCs) mediante la reprogramación celular, se han encontrado sobreexpresados en tumores agresivos y pobremente diferenciados, sugiriendo que algún tipo de desdiferenciación ocurre durante la formación del tumor (13) (14). Dos de estos factores (Myc y Klf4) son conocidos oncogenes, lo que resalta aún más el paralelismo entre estos dos procesos(15)(16).
Otros ensayos han sugerido que Sox2 está involucrado en la invasión y metástasis en muestras de neoplasia intraepitelial pancreática y en la carcinogénesis gástrica y de próstata. La expresión de Sox2 se ha observado en un alto número de carcinomas del tipo basal-like, vinculándolo a un fenotipo menos diferenciado y por otro lado, se ha propuesto que la reactivación de Sox2 representa un estadio temprano en la iniciación tumoral(17).
Oct4 se ha propuesto como marcador de tumores de líneas germinales y su expresión en células somáticas adultas resulta en falta de diferenciación, generando crecimiento displásico en tejidos epiteliales(18). La expresión de Nanog se ha asociado a peor pronóstico en carcinoma oral escamoso, y su expresión junto con Oct4 también lo es en carcinoma nasofaríngeo(19).
> Vía del mevalonato y HMGCR en cáncer
La reactivación de la síntesis de novo de lípidos, y en particular de colesterol, en tumores es un proceso que ha ganado importancia en los últimos años(20). La vía del mevalonato (MVA) es una vía anabólica esencial en las células, que utiliza acetil-CoA para producir esteroles e isoprenoides(21). Uno de los productos finales de esta vía es el colesterol, componente fundamental de las membranas celulares en vertebrados. Además, el colesterol es el precursor de múltiples biomoléculas de importancia biológica, como el ácido bílico, las hormonas esteroideas y los oxiesteroles(22).
Por otro lado, el colesterol es fundamental para la biogénesis de las balsas lipídicas (lipid rafts), ensamblados de lípidos y proteínas presentes en las membranas celulares, que están involucrados en el tráfico celular, transducción de señales y polarización celular. Esta vía también sintetiza dolicol y ubiquinona, moléculas involucradas en la glicosilación de proteínas, en el transporte de electrones en la mitocondria, y que cumplen funciones antioxidantes(23). Por otro lado, algunos isoprenoides sintetizados en esta vía son esenciales para la isoprenilación de proteínas, una modificación postraduccional fundamental para cientos de proteínas de señalización que sirve para anclarlas en las membranas celulares, permitiendo su correcta localización y función(22)(23).
Datos clínicos y experimentales sugieren que la hipercolesterolemia podría ser un factor de riesgo importante en algunos tipos de cánceres, como próstata y CM(24). Por otro lado, cambios en el metabolismo del colesterol se han relacionado con el desarrollo del cáncer(25)(26). Particularmente, se ha reportado que enzimas como HMGCR, FDPS, GGPPS, FDFT1 y SQLE están sobreexpresadas en diversos tipos tumores, incluyendo melanoma, glioblastoma, pulmón, colorrectal, próstata, ovario y CM.
En general, se ha relacionado a la sobreactivación de esta vía metabólica con menor sobrevida libre de enfermedad, peor pronóstico o recurrencia más temprana(23).
Considerando el riesgo de desarrollo de cáncer o recurrencia en pacientes con obesidad o síndrome metabólico, se ha establecido una conexión entre la alteración del metabolismo lipídico y la tumorigénesis. Particularmente, los cambios en el metabolismo del colesterol observados en células tumorales incluyen no sólo la desregulación de la síntesis, sino que también alteraciones en la captación y/o eliminación.
Tanto el colesterol como elevados niveles de uno de sus receptores (LDLR) se consideran factores de riesgo, promotores del crecimiento tumoral y se asocian a peor pronóstico en cáncer de próstata, cerebro, colorrectal y CM (23). Es más, uno de los derivados del colesterol, el 27-hidroxicolesterol (27-HC), funciona como un modulador selectivo del receptor de estrógeno (ER) en tumores de CM ER+, al estimular el crecimiento tumoral y metástasis pulmonar en modelos preclínicos(27).
> Estatinas y cáncer
La disminución de la biosíntesis derivada de la vía del MVA podría resultar un interesante blanco terapéutico contra el cáncer. La inhibición de la enzima limitante de esta vía, HMGCR, se puede producir con unas drogas denominadas estatinas(28).
Los efectos antitumorales de las estatinas incluyen activación de la apoptosis y arresto del ciclo celular, inhibición de la proliferación, invasión y formación de colonias, supresión del crecimiento tumoral, angiogénesis y metástasis en diversos tipos de tumores como glioblastoma, melanona, carcinoma de células escamosas, cáncer de próstata, gástrico, pancreático, colorrectal, de ovario y CM. Los efectos citotóxicos se detectan principalmente en células tumorales, mientras que las estatinas tienen escasa o nula toxicidad en células normales(23).
Evidencia preclínica, clínica y epidemiológica ha reportado que las estatinas impiden la proliferación, aumentan la apoptosis y reducen el riesgo de recurrencia en CM(28). Estudios en CM han reportado una disminución significativa en el riesgo de recurrencia mediado por estatinas lipofílicas, más no así en estatinas hidrofílicas(29). La prescripción de estatinas antes y después del diagnóstico está asociada a disminución de la recurrencia en CM y a mayor sobrevida.
La administración de dosis altas de atorvastatina y fluvastatina, previamente a la cirugía en pacientes con CM invasivo en estadios tempranos, indujo una disminución de la proliferación y aumento de apoptosis en los tumores.
Un meta-análisis de 41 estudios que incluyeron en total más de un millón de participantes, demostró que el uso de estatinas pre y post-diagnóstico está asociado a mejor sobrevida total y sobrevida libre de enfermedad(30). Por lo tanto, los efectos clínicos individuales de las estatinas pueden depender de sus propiedades químicas, del tiempo de administración, del tipo o estadio del tumor o de otras características del paciente como edad, comorbilidad y niveles de lípidos, entre otros(23).
> Fenotipo colesterogénico y células madre tumorales
A pesar de la vasta información disponible de la vía del MVA en el desarrollo del cáncer y de las estatinas como posibles agentes preventivos y/o terapéuticos, poco se conoce acerca de su influencia específica en la población de las CMTs.
En este contexto, algunos estudios han aportado evidencia sobre el rol esencial de la vía del MVA en la regulación de la población de CMTs. Por un lado, Sharon y colaboradores demostraron que la vía del MVA se encontraba sobreactivada en CMTs de colon, al comparar cambios en los perfiles de expresión de las células crecidas en monocapa con respecto a tumoroesferas(31).
Con un enfoque similar, Ginestier y colaboradores demostraron la sobreexpresión de genes de la vía del MVA en mamoesferas derivadas de líneas celulares triple negativas en CM. Más aún, luego de un tratamiento con simvastatina, estas líneas presentaron menor proporción de CMTs, y la adición de mevalonato era capaz de restituir esta población celular(32). Fiorillo y colaboradores encontraron sobrexpresión en genes de la vía del MVA al crecer como tumoroesferas líneas celulares luminales de CM y que derivados naturales de una fruta (bergamota) eran capaces de eliminar la población de CMTs, al inhibir a HMGCR de una manera similar a como lo hacen las estatinas(33).
También se ha demostrado que el tratamiento con lovastatina inhibe propiedades de CMTs en carcinoma nasofaringeo al inducir apoptosis y arresto del ciclo celular. Además, el tratamiento con lovastatina aumentó la sensibilidad de las CMTs hacia la quimioterapia(34). En CM, se aisló la población de CMTs en una línea luminal y se encontró una mayor apoptosis que en la línea parental luego de un tratamiento con simvastatina (35), y por otro lado se comprobó que el tratamiento con lovastatina dismminuyó la población de CMTs en líneas triple negativas. La reversión de estos efectos con la adición de mevalonato sugieren que los efectos anti-CMT de la lovastatina podrían atribuirse, al menos en parte, a la inhibición de HMGCR(36).
Por otro lado, se encontró que líneas celulares de CM resistentes a drogas estaban enriquecidas en CMTs, generando en consecuencia tumores más agresivos, y que el tratamiento con simvastatina era capaz de eliminarlas específicamente(37). Uno de los tratamientos estándares en osteosarcoma es el uso de adriamicina.
Estudios recientes han reportado que esta droga induce propiedades de CMT y promueve el potencial metastásico en células de osteosarcoma al activar el oncogen KLF4. Interesantemente, el tratamiento con estatinas induce la pérdida de las propiedades de CMTs vía inhibición de KLF4(38).
Rennó y colaboradores demostraron los efectos inhibitorios de simvastatina sobre las CMTs en un modelo in vivo de carcinogénesis mamaria. Especificamente, encontraron que el tratamiento con esta droga, una vez que el tumor ya se ha establecido, inhibe el crecimiento tumoral, modula la heterogeneidad morfológica y disminuye la expresión de características asociadas a CMTs en mama(39).
En resumen, estos resultados indican, por un lado, que la vía del MVA está involucrada en la generación y mantenimiento de estados de CMTs en distintos tumores y por otro, que las estatinas podrían utilizarse para la eliminación selectiva de esta subpoblación. Por lo tanto, estas drogas podrían utilizarse como adyuvantes en terapias convencionales.
> Relevancia del proyecto
Las CMTs se definen por su habilidad para autorrenovarse y recapitular la heterogeneidad completa del tumor en cultivo y en xenoinjertos. Son altamente tumorigénicas y se las implica directamente en la resistencia a quimioterapia y radioterapia, lo que les confiere una gran importancia como dianas terapéuticas potenciales en cáncer, y en particular en CM.
Dada la baja frecuencia e inestabilidad in vivo de las CMTs, hay un interés creciente en la generación de modelos de CMTs in vitro, ya que podrían utilizarse para recrear el proceso de transformación oncogénica y los estadios tempranos del desarrollo tumoral. Esto proveería herramientas de valor para estudiar los mecanismos regulatorios involucrados en la obtención y mantenimiento de estados CMTs y proveer plataformas para el desarrollo de drogas específicas(19). En este contexto, en el presente trabajo se propone el uso de distintas estrategias para modelar in vitro estados de CMT en CM con el fin de dilucidar el rol de la enzima HMGCR durante este proceso.
| Materiales y Métodos |
> Sistema CRISPR/Cas9
CRISPR/Cas9 es un sistema inmune adaptativo presente en bacterias y Archaeas que las protegen de infecciones virales(40). El término CRISPR hace referencia a loci presentes en los genomas de estos microorganismos, que contienen secuencias con repeticiones palindrómicas cortas, separadas por un “DNA espaciador” proveniente de exposiciones previas a un virus.
Los genes Cas (CRISPR-associated system) codifican para las nucleasas encargadas de cortar los elementos génicos exógenos, reconocidos por las secuencias espaciadoras(40)(41).
Este sistema ha sido adaptado para su uso en el laboratorio como método de edición génica. En este proyecto se utilizó una modificación del sistema original conocido como “CRISPRon”, que consiste en un sistema activador de la transcripción con dos componentes.
Uno de ellos es una proteína Cas9 fusionada a un dominio activador (VP160), cuya unión a la secuencia blanco se basa en la complementariedad de bases. Esta proteína presenta mutaciones que la inhabilitan para cortar el DNA pero que aún es capaz de dirigirse hacia la zona blanco, guiada por el segundo componente del sistema, los RNA guías. En el sistema CRISPRon, los RNA guías son diseñados para dirigir a la Cas9 al promotor del gen de interés(42)(43).
> Plásmidos
Se utilizó un sistema CRISPR-Cas9 para activar el gen HMGCR humano. Este sistema está compuesto por un plásmido guía (pSPgRNA, Addgene # 47108), en el cual se clonó la secuencia de interés y un plásmido efector que codifica para la nucleasa Cas9 inactivada, junto con un dominio activador (pAC94-pmax-dCas9-VP160-2A-puro, Addgene # 48226). Ambos plásmidos fueron cedidos por el Dr. Federico Pereyra Bonnet del Instituto de Ciencias Básicas y Medicina Experimental del Hospital Italiano (ICBME).
> Transformación de bacterias, purificación y verificación del DNA plasmídico
Se transformaron bacterias competentes de la cepa DH5α de Escherichia coli con los plásmidos utilizados en este trabajo. En todos los casos, se realizó la transformación mediante shock térmico. Para ello, se incubó 1 µg del plásmido en 100 µL de bacterias competentes de la cepa mencionada por 30 minutos en hielo, seguido de 90 segundos a 42°C y 5 minutos en hielo nuevamente. Después, se agregaron 800 µL de medio Luria Bertani (LB) sin antibiótico y se incubó por 45 minutos a 37°C.
Luego de una centrifugación de 5 minutos a 1.000 rpm para concentrar las bacterias, las mismas fueron sembradas en placas de Petri con LB y el antibiótico correspondiente (kanamicina 30 µg/mL para el plásmido activador y ampicilina 100 µg/mL para el plásmido guía). Las placas fueron incubadas overnight (ON) en estufa a 37°C. Al día siguiente, se seleccionó una colonia y se la sembró en 3 mL LB líquido con antibiótico y se incubó nuevamente ON a 37°C.
Este cultivo líquido se utilizó para obtener un stock de glicerol y para inocular un volumen mayor de medio (30 mL) con antibiótico que permitió amplificar el cultivo de bacterias transformadas. Se realizó una “midiprep” utilizando un kit de Quiagen (Quiagen Plasmid Midi Kit, cat #12143, Thremo) para lisar las bacterias transformadas y purificar el DNA de los plásmidos siguiendo las instrucciones del fabricante. Finalmente, se verificaron los plásmidos por digestión enzimática.
> Generación del Plásmido Guía del sistema CRISPR
• Diseño y síntesis de sondas: Se utilizó una herramienta online gratuita disponible en la página http://crispr.mit.edu para seleccionar cinco sondas con homología a la región proximal del promotor del gen HMGCR humano. Las sondas fueron sintetizadas por Genbiotech.
• Clonación de sondas: Se utilizó el protocolo de clonación de sondas del laboratorio del Dr. Zhang, disponible en la página http://www.genomeengineering.org/crispr/. Brevemente, se fosforilaron e hibridizaron cada par de sondas utilizando la enzima T4 PNK (cat # M0201S, NEB) a 37°C por 30 min y luego a 95°C por 5 min, seguido de una disminución progresiva de 5°C cada 5 min hasta llegar a los 25°C.
Por otro lado, por cada clonado a realizar, se digirió 1 ug de plásmido guía con la enzima BbsI (cat# ER1011, Thermo) a 37°C ON, la cual posee dos sitios de corte en la secuencia de interés (Figura 1). Luego, se agregó 1 µL de FastAP (cat # EF0654, Thermo) y se incubó 15 min a 37°C, seguida de una inactivación de 5 min a 75°C. Posteriormente, se sembraron las muestras en un gel de agarosa al 1% (P/V) con bromuro de etidio y se realizó una corrida electroforética con buffer TAE (Tris- Acetato 40 mM pH = 8; EDTA 1 mM) a 80 V durante 90 minutos. Se purificaron las bandas del gel utilizando un kit comercial (Illustra GFX PCR DNA and Gel Band Purification Kit, cat # 209034-70, GE Healthcare), siguiendo las instrucciones del fabricante.
Finalmente, se realizó la ligación del plásmido guía digerido con los dúplex de oligonucleótidos fosforilados e hibridados utilizando la enzima T4 Ligasa (cat # EL0014, Thermo) durante 10 min a temperatura ambiente, seguido de una incubación de 1 hora a 4°C.
• Transformación de bacterias y purificación de los plásmidos: Se transformaron bacterias competentes de la cepa DH5α con el plásmido guía clonado con cada par de sondas mediante la técnica de shock térmico y se extrajo el DNA plasmídico como se describió previamente.
• Verificación por secuenciación: Se seleccionaron dos colonias de bacterias transformadas con cada par de sondas clonadas, para secuenciar utilizando los servicios de Macrogen. Con los programas informáticos FinchTV y MEGA se determinó la presencia y la orientación del inserto en los clones evaluados.
 Figura 1: Fragmento de la secuencia del plásmido guía donde se realiza la digestión enzimática con BbsI. Los sitios de corte se resaltan con triángulos rojos. Tomado de Genome Engineering Toolbox, Zhang Lab.
Figura 1: Fragmento de la secuencia del plásmido guía donde se realiza la digestión enzimática con BbsI. Los sitios de corte se resaltan con triángulos rojos. Tomado de Genome Engineering Toolbox, Zhang Lab.
> Cultivo celular
Se utilizó la línea celular derivada de CM MCF-7, la línea comercial de células madre embrionarias WA-09, obtenidas del WiCell (Wisconsin, USA) por el LIAN del Instituto Fleni, y la línea de células madre con pluripotencia inducida (Induced Pluripotent Stem Cells, iPSCs) FN2.1, esta última, generada y caracterizada en dicho laboratorio(44).
El panel de CMTs derivadas de gliomas humanos utilizado en este trabajo fue obtenido y caracterizado por el Dr. Videla- Richardson en el Instituto Fleni(45). Se utilizaron frascos de cultivo (T25 y T75), placas de cultivo de Petri (p35, p60 y p100) y placas multipocillo (p96, p24, p12 y p6) de las marcas Corning (Corning, NY, EEUU.) y JetBiofil (Guangdong, China). Todas las líneas se mantuvieron en estufa de cultivo a 37°C en atmósfera de CO2 5%. Los medios de cultivo y suplementos utilizados para cada línea celular se detallan en la Tabla 1.
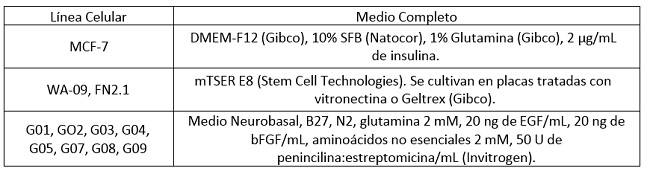 Tabla 1: Composición de los medios de cultivo utilizados para cada línea celular (SFB, Suero Fetal Bovino).
Tabla 1: Composición de los medios de cultivo utilizados para cada línea celular (SFB, Suero Fetal Bovino).
> Transfección de líneas celulares con el sistema CRISPR.
Se partió de 350.000 células sembradas en placas de 6 pocillos (p6). Aproximadamente, al llegar a 80% de confluencia, se transfectaron las células con los plásmidos del sistema CRISPR. Se utilizó el reactivo de transfección FuGENE (cat #E2311, Promega), siguiendo las instrucciones del fabricante. Brevemente, se utilizó la relación recomendada 3:1 de reactivo de transfección con respecto a la cantidad de plásmido.
Para los controles de transfección se utilizó 50% de plásmido activador y 50% de plásmido guía vacío, es decir, sin las secuencias que codifican para las diferentes sondas. Para las células con inducción de la expresión de HMGCR se utilizó 50% de plásmido activador y 50% de plásmido guía clonado (10% correspondiente a cada par de sondas).
Se colocaron los plásmidos y el FuGENE en tubos, los cuales fueron mezclados utilizando vórtex por 10 segundos e incubados a temperatura ambiente (T°amb) por 15 minutos. Luego, se colocaron 150 µL de esta mezcla en pocillos de p6, que ya tenían 3 mL de medio fresco. Las placas se incubaron en estufa hasta el momento de procesar las muestras.
> Extracción de RNA y Retrotranscripción (RT).
Se partió de células sembradas en placas de 6 o 12 pocillos (p6 o p12). Se descartó el medio de las mismas, se agregaron 500 µL de Tri Reagent (cat # TR118, Genbiotech) y se utilizó una espátula para liberar el RNA. El homogeneizado se colocó en un tubo y luego de 5 minutos de incubación a temperatura ambiente, se agregaron 100 µL de cloroformo.
Los tubos fueron agitados con vórtex por 15 segundos e incubados 3 minutos a temperatura ambiente. Luego fueron centrifugados a 12.000 rpm por 15 minutos a 4°C y la fase acuosa fue transferida a un tubo nuevo. Se agregaron 250 µL de isopropanol a cada tubo y se incubaron ON a -20°C. Luego, se realizó una centrifugación a 12.000 rpm por 10 minutos a 4°C para precipitar el RNA y se removió el sobrenadante.
Se lavó el pellet de RNA resultante con 500 µL de etanol al 75% (V/V), seguido de una centrifugación de 5 minutos a 7.500 rpm. Este paso de lavado se realizó dos veces. Se procedió a secar los pellets en una placa térmica a 58°C por 5 minutos y luego se resuspendió el RNA en agua a la misma temperatura por 10 minutos. Finalmente, se cuantificó el RNA utilizando el equipo Nanodrop y se lo conservó a -70°C.
El DNAc se sintetizó a partir del RNA total mediante retrotranscripción utilizando el kit Easy Script Reverse Transcriptase (cat # AE101, TransGen). Brevemente, en un tubo se colocó el RNA, 1 μL de Oligo(dT) y agua hasta llegar a un volumen final de 10,5 μL.
Los tubos fueron incubados 5 minutos a 65°C y luego 2 minutos en hielo. En otro tubo se preparó la Master Mix (MM) compuesta por dNTPs 2,5mM o 10mM (cat # AD101-01 y AD101-11, TransGen) y la enzima retrotranscriptasa con su respectivo buffer 5x. Posteriormente, se agregaron 9,5 μL de la MM a cada tubo de muestra. Por último, se incubaron las reacciones por 30 minutos a 42°C, seguido de una inactivación de 5 segundos a 85°C. En cada reacción se realizaron controles sin enzima, para corroborar que no haya amplificación genómica. El DNAc se conservó a -20°C.
> Reacción en Cadena de la Polimerasa en tiempo real (RT-qPCR).
La medición de los niveles de RNAm se realizó por PCR en tiempo real con el equipo Step One Plus (Applied Biosystems). Las reacciones se llevaron a cabo utilizando las mezclas comerciales PowerUp™ SYBR™ Green Master Mix (cat # A25742, Thermo) y TransStart® Green qPCR SuperMix (cat # AQ101, TransGen).
A las mezclas se agregaron cebadores de interés (0,2-0,4 µM), muestra y agua en un volumen final de 20 μL. Los ensayos se realizaron por triplicado y se incluyeron en cada reacción controles negativos de PCR (controles de agua y de amplificación de DNA genómico).
Los niveles de expresión de los transcriptos de los genes evaluados fueron estimados mediante el método de ΔΔCt, utilizando GAPDH o RPL7 como gen endógeno para la normalización. Se utilizaron muestras de referencia para la relativización en cada análisis. Las secuencias de cada par de cebadores utilizados se detallan en la Tabla 2.

Tabla 2: Secuencias y temperatura de hibridación de los cebadores utilizados en este trabajo.
> Ensayos de formación de colonias en agar blando (Soft-agar assay)
Se realizaron ensayos de formación de colonias en agar blando para estudiar el efecto de la sobreexpresión de HMGCR sobre la tumorigenicidad de la línea MCF-7. Esta técnica permite cuantificar la cantidad de células con la capacidad de crecer de forma independiente de anclaje, una característica particular de células cancerígenas. Se trataron placas de 12 pocillos con 1 mL/pocillo de una solución de agarosa de bajo punto de fusión (low melting point, LMP) UltraPURE (cat # 5517UB, Gibco) al 2% (P/V) en DMEM-F12 completo.
Las placas se dejaron solidificar 1 hora a T° amb. Las células fueron resuspendidas en medio completo con 1% de agarosa LMP y se sembraron 10.000 células en cada pocillo, previamente tratado con la solución de agarosa al 2%. Se incubaron a 37°C durante 1 hora y, luego, se agregó 1 mL de DMEM-F12 completo. Se dejaron incubando a 37°C, agregando 250 μL de medio completo cada 2-3 días.
En cada ensayo, se sembraron dos pocillos de cada línea. Luego de 30 días, las colonias formadas se fijaron y tiñeron con una solución de MeOH al 40% (V/V) y cristal violeta al 0,05% (P/V) en PBS durante 1 hora a - 20°C. Se visualizaron y tomaron fotografías en cada pocillo con un microscopio invertido de contraste de fase (Olympus CKX-41).
> Ensayos de viabilidad celular.
Se realizaron ensayos de reducción de 3-(4,5-dimetiltiazol-2-il)-5-(3-carboximetoxifenil)-2-(4- sulfofenil)-2H-tetrazolio (MTS) con el kit CellTiter 96® AQueous One Solution Cell Proliferation assay (cat # G3582, Promega), para estudiar la viabilidad celular en distintas condiciones. Las células se sembraron en placas de 96 pocillos (p96) en un volumen de 200 µL de medio.
Cada línea se sembró por triplicado. Se incubaron a 37°C y al momento de realizarse los experimentos, se descartó el medio y se agregaron 50 μL de DMEM-F12 base fresco, por pocillo. Además se colocó este volumen de medio en tres pocillos vacíos, que se utilizaron como blancos de reacción.
En oscuridad, se agregaron 10 μL de la solución de MTS a todos los pocillos. Las placas se incubaron en oscuridad a 37°C por 4 h, midiendo la absorbancia en lector de ELISA a 490 nm cada hora. Posteriormente, se calculó la viabilidad celular y se relativizó al control (100%).
> Ensayos de adhesión celular
Se sembraron 60.000 células por cada pocillo en placas p96 en un volumen de 200 µL de medio completo. Cada línea se sembró por triplicado. Se incubaron las placas a 37°C y 5 %CO2 por 90 minutos. Luego se descartó el medio, se hicieron dos lavados con 100 μL de PBS/pocillo (PBS 1x) y se fijaron las células con 100 μL de metanol (MeOH) a T° ambiente por 10 minutos.
Se descartó el MeOH y se lavaron los pocillos con 100 μL de PBS. Luego, se tiñeron con Cristal Violeta (CV) al 0,5% durante 10 minutos a T° ambiente. Este paso, y los posteriores, se realizaron a su vez en tres pocillos vacíos y fueron utilizados como blancos de reacción. Se realizaron dos lavados con 100 μL de agua destilada para eliminar el exceso de colorante.
Luego, se resuspendió el CV que se había incorporado a las células con 60 μL de solución de MeOH al 10% y ácido acético al 5% (V/V) en agua destilada. Finalmente, se midió la absorbancia en lector de ELISA a 620 nm y se calculó la adhesión celular, relativizando los datos al control (100%).
> Ensayos de migración celular (insertos).
Se evaluó el efecto de la sobreexpresión de HMGCR sobre la capacidad migratoria de células MCF-7 utilizando insertos (cat # TCS003024, Biofil). Para ello, se colocaron 500 μL de medio completo en cada pocillo de placas p24 y, posteriormente, se colocaron los insertos. A las 48 horas de realizarse la transfección con el sistema CRISPR, se sembraron 15.000 células resuspendidas en 200 µL de DMEM-F12 base en la parte superior del inserto.
Las placas se incubaron a 37°C por 15 horas y, luego, se fijaron en MeOH por 10 minutos a T° ambiente. Posteriormente, se tiñeron con DAPI y se lavaron con agua destilada. Las células que no migraron (que se encontraban en la parte superior del inserto) fueron retiradas con un hisopo húmedo. Las membranas se dejaron secar y, luego, se tomaron imágenes con microscopio invertido de contraste de fase (Olympus CKX-41). Se contó la cantidad de células que migraron en cada línea y se relativizó a la línea control (100%).
> Extracción de proteínas y Western Blot.
Se utilizó la técnica de Western Blot (WB) para detectar y comparar los niveles de proteínas en los lisados de las líneas celulares del sistema CRISPR. Obtención de los lisados celulares: Se partió de células sembradas en placas p12, a las que se les descartó el medio y se agregaron 50 μL de buffer de lisis (Tris-HCl 60 mM pH 6,8 y SDS al 1% (P/V) en H20d). Se utilizaron espátulas para liberar las proteínas. Se recuperaron los lisados en tubos tipo Eppendorf y se hirvieron por 3 minutos. Luego se agitaron las muestras por 20 seg y se hirvieron por 7 minutos nuevamente.
Se centrifugaron los tubos a 13.000 rpm por 10 minutos y se pasaron los sobrenadantes a tubos nuevos. Las muestras se cuantificaron con nanodrop. Corrida electroforética, transferencia y detección: Se sembraron cantidades iguales de proteína total (60-80 μg), mezclada con el volumen correspondiente de buffer de carga 6X (Tris 0,5M, SDS 1 g, glicerol 3 mL, azul de bromofenol 1 g, β- mercaptoetanol 680 μL, pH = 6,7) en cada calle de geles desnaturalizantes SDS-PAGE (12%) de poliacrilamida (acrilamida cat # 15512-023, Invitrogen; N,N´-metilenbisacrilamida cat # 15516-024, Invitrogen) (espesor 1,5 mm). Se sembró también un marcador de peso molecular (cat #161-0373, Bio-Rad) en cada gel.
Las proteínas se separaron por electroforesis: inicialmente 30 minutos a 60 V y luego 1:45 h a 100 V y se transfirieron a membranas de nitrocelulosa (cat # 10600002, Amersham) durante 1:45 h a 80 V. Las membranas fueron bloqueadas con seroalbúmina bovina (SAB, cat # 0055K, Sigma-Aldrich) en buffer Tris salino con Tween 20 (TBS-T 0,05%) por 1 hora y, luego, incubadas con los diferentes anticuerpos primarios overnight (ON) a 4°C con agitación.
Luego, se lavaron las membranas y se incubaron con el correspondiente anticuerpo secundario acoplado a peroxidasa (HRP) durante 1:30 horas. Se lavaron 3 veces con TBS-T y se procedió a la detección. Los complejos con el anticuerpo se visualizaron usando quimioluminiscencia (ECL; GE Healthcare) con el equipo G:BOX (Syngene, UK). La intensidad de las bandas fue medida utilizando el programa informático especializado ImageJ (NIH, Bethesda, MD, USA). Se utilizó tubulina como control de carga. Anticuerpos y diluciones utilizadas: HMGCR (1/300, cat #sc-271595, Santa Cruz Biotechnology), tubulina (1/5000, cat # ab52623, Abcam), anti-rabbit-HRP 1/6000 (cat # A0545, Sigma) y anti-mouse-HRP (1/4000, cat #A4416,Sigma).
> Generación y cuantificación de mamoesferas (ELDA).
El ensayo de mamoesferas se realizó basado en modificaciones(8) del protocolo original de Dontu y col.(46). Con este propósito, se trataron placas de cultivo p6 y p96 con poli 2- hidroxietilmetacrilato (cat # P3932, Sigma) para evitar la adhesión celular.
Por cada pocillo de placa p6, se sembraron 3.000 células/mL en 3 mL de medio de esferas completo, compuesto por DMEM/F12 enriquecido con 2% de B27, 1% de glutamina, 0,1% de metilcelulosa, 20 ng EGF/µL (Factor de Crecimiento Epidérmico, EGF) y 20 ng bFGF/µL (Factor de Crecimiento de Fibroblastos básico, bFGF).
Las células se dejaron crecer durante 7-10 días, agregando 0,5 mL de medio de esferas y factores de crecimiento cada 3-4 días. Para una cuantificación más precisa de las mamoesferas se utilizó una modificación del ensayo de dilución límite (Extreme Limiting Dilution Assay, ELDA), que permite calcular mediante análisis estadístico la frecuencia de formación de mamoesferas por número de células sembradas, asumiendo que se trata de esferoides clonales y no multicelulares(47). Para estos ensayos se utilizaron placas p96 tratadas con poli-2- hidroxietilmetacrilato.
Se sembraron 200 µL de suspensiones celulares con concentraciones decrecientes de células por cada pocillo (300, 100, 30 y 10 células). Luego se cultivaron las células por 7-10 días y se contó en el microscopio la cantidad de eventos positivos, representados por la presencia de, al menos, una esfera en cada pocillo. Finalmente, se ingresó el número de células (Dose), la cantidad de pocillos ensayados en cada condición (Tested) y la cantidad de eventos positivos (Response) en la herramienta bioinformática disponible online en el Walter and Eliza Hall Institute of Medical Research Bioinformatics Division (http://bioinf.wehi.edu.au/software/elda) para la cuantificación de unidades formadoras de esferas, es decir, para calcular la frecuencia de células madre o progenitoras en las líneas estudiadas (Figura 2).
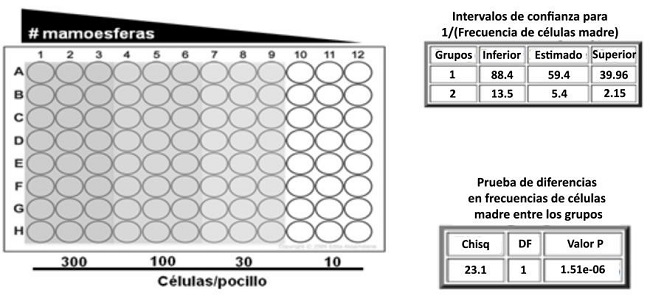
Figura 2: Ensayo de ELDA. Se muestra el diseño de la placa p96 y los números de células sembradas. El análisis da como resultado la frecuencia de células madre o progenitoras (1/stem cell frequency).
> Determinación de la población CD44+/CD24low/- y CD133+ por citometría de flujo
Luego de la transfección de células MCF-7 con el sistema CRISPR/CAS9 para HMGCR y con los correspondientes controles, se colocaron 150.000 células en un tubo, se centrifugaron y se lavaron con PBS. Las células se volvieron a centrifugar, se descartó el sobrenadante y se resuspendieron en 500 μL de PBS. Se centrifugaron las muestras a 400 xg por 5 minutos a 4°C, se descartó el PBS y se resuspendieron las células en 20 μL de solución de bloqueo (suero de cabra al 10% en PBS) por 30 min a T° amb, en oscuridad. Luego, se agregaron 20μL de las diluciones correspondientes de anticuerpos y se incubaron los tubos durante otros 30 minutos en oscuridad, en hielo.
Las diluciones utilizadas fueron: para CD44 (cat # 103012, Biolegend) 1/80 y para CD24 (cat # 311106, Biolegend) 1/100, en solución de bloqueo. Se centrifugaron las muestras a 400 xg por 5 minutos a 4°C, se descartó el sobrenadante y se fijaron las células con 100 μL de PFA al 4% por 30 minutos, en oscuridad. Luego de otra centrifugación a 400 xg por 5 minutos a 4°C se descartó el sobrenadante, se resuspendieron las células en 100 μL de PBS y se almacenaron a 4°C hasta la adquisición de los datos. Para la detección de la población CD133+ se centrifugaron las muestras a 300 xg por 10 minutos a 4°C, se descartó el PBS y se resuspendieron las células en 80 μL de buffer (EDTA 2 mM, BSA 0,5% en PBS). Se agregaron 2 μL de anticuerpo CD133 (cat # AC133, VioBright ™ 515) a cada tubo y se incubó por 10 minutos a 4°C en oscuridad, quedando una dilución 1/50.
Se agregaron 500 μL de buffer a cada tubo, se centrifugó a 300 xg por 10 minutos a 4°C y se descartó el sobrenadante. Se fijaron las células con 200 μL de PFA al 2% por 10 minutos a 4°C. Luego, se agregaron 500 μL de buffer a cada muestra, se centrifugó y se descartó el sobrenadante. Se resuspendieron las células en buffer. En todos los casos, se pasaron las muestras por el citómetro de flujo (BD FACScanto™ II). Se utilizaron los programas especializados FacsDiva y FlowJo para la adquisición y análisis de los datos, respectivamente.
| Estadística |
Se utilizó el programa informático especializado GraphPad Prism® versión 6 (GraphPad Software Inc., CA) para realizar los análisis estadísticos. Los resultados se presentan como la media
± el error estándar de la media (EEM) del número de experimentos independientes indicados en cada caso. Los grupos experimentales se compararon utilizando los tests estadísticos no paramétricos Mann-Whitney o Kruskal-Wallis, para dos o más grupos, respectivamente. La diferencia significativa se evaluó utilizando test de dos colas. Se consideró que había una diferencia significativa cuando el valor de p era menor a 0,05.
| Resultados |
> Diseño y síntesis de sondas
Se utilizó la herramienta informática Genome Engineering Toolbox (MIT, Cambridge, MA) del laboratorio Zhang para diseñar sondas específicas para la región proximal del promotor del gen HMGCR humano. Este programa evalúa la homología entre cada sonda generada y la zona de interés, notificando el número de mismatches. Además, evalúa la posibilidad de que se produzcan apareamientos con otras regiones del genoma (off-target sites). Estos datos se informan con un número (score), de modo que el valor 100 representa la sonda con menor probabilidad de apareamiento no deseado.
En la Figura 3.A pueden observarse los resultados obtenidos con esta herramienta, detallando las posibles sondas para la región de interés y los datos relevantes para su evaluación. Con esta información se seleccionaron cinco sondas, también denominadas RNAs guías (gRNAs) dirigidos al promotor del gen HMGCR humano. En esta figura también se detallan las secuencias de las cinco sondas elegidas y sintetizadas, y además se muestra un esquema representativo de las zonas blanco de cada sonda en el promotor de HMGCR (Figura R3.B).
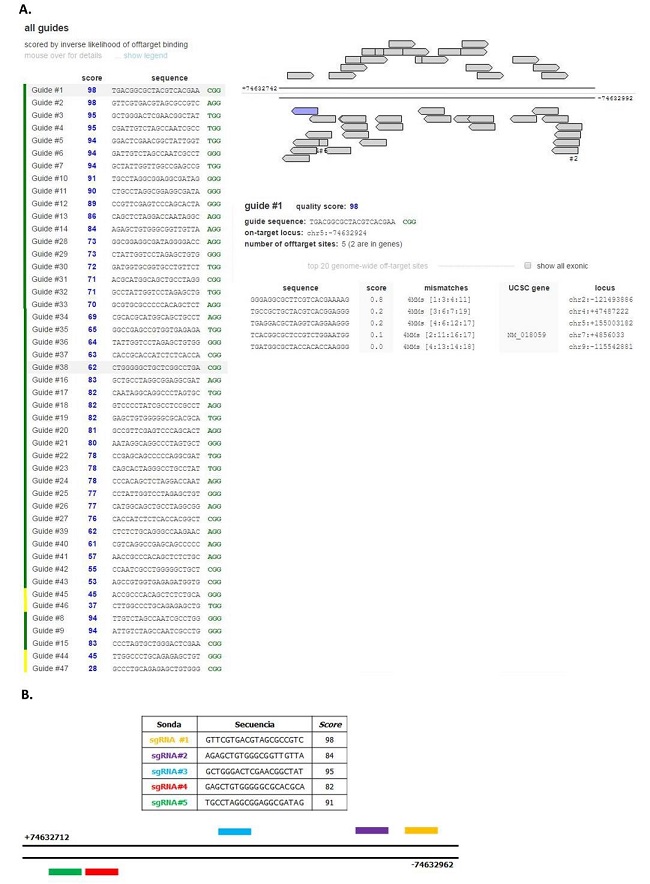
Figura 3: Sondas de la región proximal del promotor de HMGCR. A. Resultado obtenido con la herramienta Genome Engineering Toolbox del Zhang Lab, donde se detallan sondas posibles para la región de interés. B. Secuencias y Score de las cinco sondas elegidas y esquema representativo de las zonas blanco de una de ellas en el promotor del gen HMGCR humano.
> Clonación de las sondas en el plásmido guía y verificación por secuenciación
Como se mencionó previamente, se utilizó el protocolo de clonación recomendado por el laboratorio del Dr. Zhang. Brevemente, se digirió enzimáticamente el plásmido guía utilizando la enzima BbsI y, luego de migrar en gel de agarosa, la banda lineal del plásmido guía digerido fue purificada con un kit comercial.
Posteriormente, las sondas diseñadas se fosforilaron, hibridizaron y se realizó la ligación con el plásmido guía digerido. Finalmente, se transformaron bacterias competentes con los plásmidos clonados con cada sonda y se seleccionaron dos colonias representativas de cada placa para secuenciar.
Con el programa informático FinchTV se determinó la presencia y orientación del inserto en los clones evaluados, verificando de esta forma el correcto funcionamiento de la clonación. Por otro lado, se utilizó el programa MEGA para alinear y comprobar las secuencias del plásmido guía digerido y de los plásmidos clonados con las sondas (Figura 4).
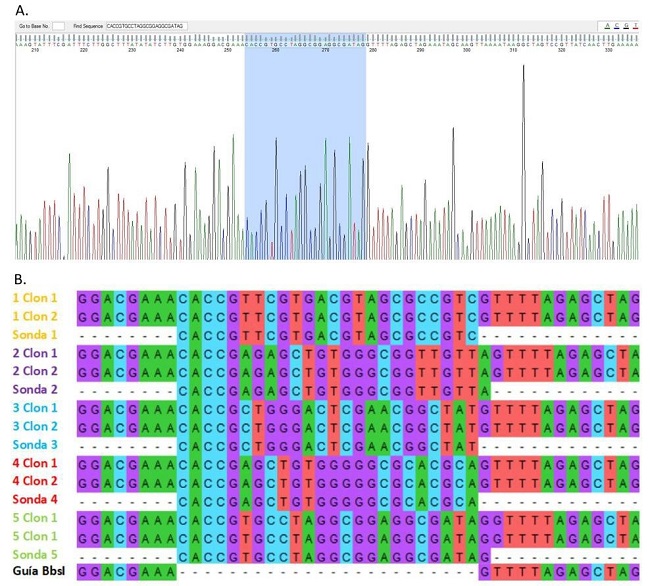
Figura 4: Verificación de la clonación por secuenciación. A. Análisis realizado con la herramienta bioinformática FinchTV, donde se muestra la secuencia del inserto y la correcta orientación del mismo. B. Alineamiento de secuencias realizado con el programa MEGA7, se incluye la secuencia del plásmido guía digerido, de las sondas sintetizadas y de dos clones por cada par de sondas.
> Inducción del fenotipo HMGCRon en la línea celular MCF-7
Luego de verificar la clonación del plásmido guía, se transfectó la línea MCF-7 con el sistema CRISPRon. A las 48 horas post-transfección se evaluaron los niveles de HMGCR por RT-qPCR, confirmando que el sistema CRISPRon aumenta la expresión de HMGCR total en las células tratadas (MCF-7/CR) con respecto al control de transfección (MCF-7/TC) (Figura 5.A).
Además, se evaluaron los niveles de expresión de HMGCR a distintos tiempos, encontrando poca variación entre las 48 y 96 horas post-transfección. Este resultado permitió mayor versatilidad para planificar los experimentos, ya que se contó con un mayor lapso de tiempo para realizar los ensayos de caracterización de este sistema (Figura 5.B).
También se midieron a nivel transcripcional los niveles de expresión de las isoformas de HMGCR (48 horas post-transfección), una de tamaño completo (HMGCR-FL) y una con una exclusión del exón 13 (HMGCR-del13), encontrándose ambas elevadas en las células MCF-7/CR (Figura 5.C). Además, se observó un incremento de los niveles proteicos de HMGCR a este tiempo, indicando que el sistema activador CRISPRon utilizado efectivamente induce la expresión del gen blanco, tanto a nivel transcripcional como proteico (Figura 5.D).
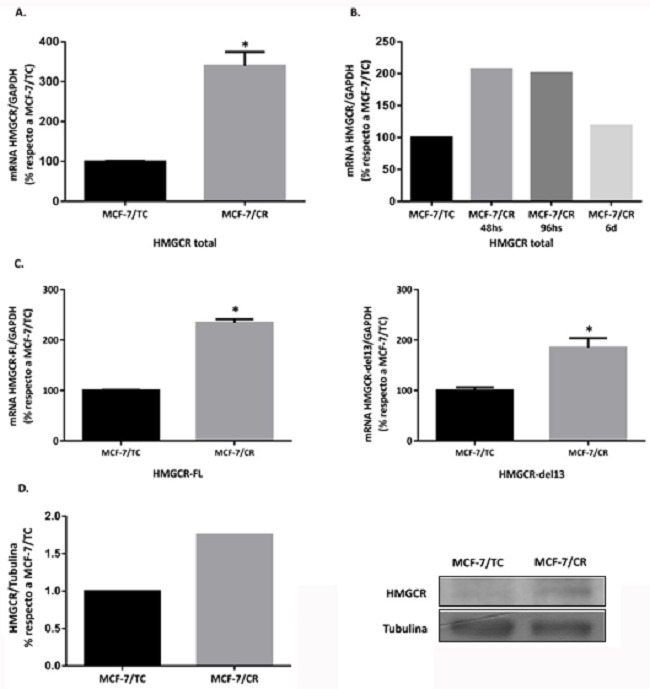
Figura 5: Inducción del fenotipo HMGCRon luego de la transfección con el sistema CRISPRon
A. Análisis de expresión de HMGCR total a 48 horas post-transfección. B. HMGCR total a distintos tiempos post-transfección (48h, 96h y 6 días). C. Isoformas de HMGCR (HMGCR-FL y HMGCR-del13) a 48 horas post-transfección Test Mann-Whitney (*p<0.05 vs. MCF-7/TC). Se muestra la media ± EEM de 3 experimentos independientes. D. Western Blot para la proteína HMGCR a 48 h post-transfección (n=1).
> Características asociadas a células madre
Buscando estudiar la relación entre HMGCR y propiedades de células madre se realizaron ensayos funcionales para la detección de ciertas características presentes en células madre/progenitoras, como la formación de mamoesferas. Para cuantificar la cantidad de “unidades formadoras de mamoesferas” realizamos Ensayos de Dilución Límite (ELDA). Al comparar las células MCF-7/TC y MCF-7/CR, no se observaron diferencias en la formación de mamoesferas en cuanto a morfología o tamaño, pero sí se puso apreciar una mayor tendencia a formar esferas en las células MCF-7/CR, sugiriendo que poseen un mayor número de células madre o progenitoras (Figura 6).
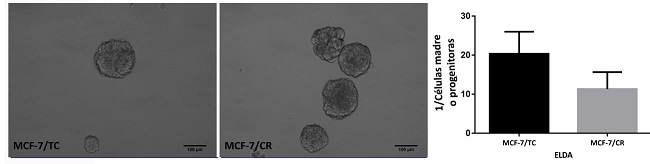 Figura 6: Ensayos de mamoesferas. Imágenes representativas de mamoesferas obtenidas de las células MCF-7/TC y MCF-7/CR y cuantificación de unidades formadoras de esferas para ambas líneas (ELDA). Test Mann-Whitney. Se muestra la media ± EEM de 3 experimentos independientes.
Figura 6: Ensayos de mamoesferas. Imágenes representativas de mamoesferas obtenidas de las células MCF-7/TC y MCF-7/CR y cuantificación de unidades formadoras de esferas para ambas líneas (ELDA). Test Mann-Whitney. Se muestra la media ± EEM de 3 experimentos independientes.
Luego, se estudiaron marcadores de células madre tumorales en CM, como la proporción de células CD44+/CD24-/low y CD133+ por citometría de flujo. En ambos casos se encontró una tendencia a aumentar las poblaciones de interés en las células MCF-7/CR con respecto al control de transfección MCF-7/TC (Figura 7).
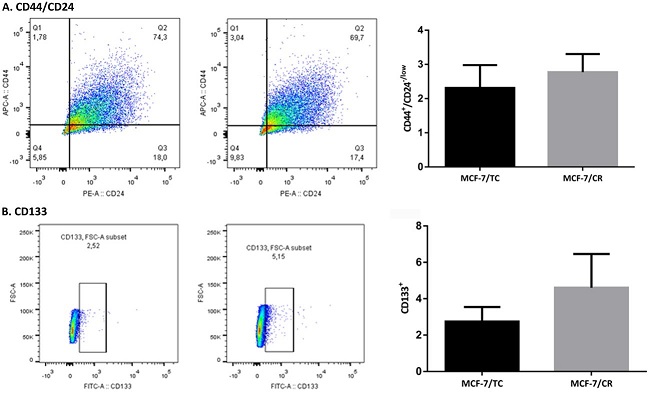
Figura 7: Subpoblaciones CD44+/CD24-/low y CD133+ por citometría de flujo A. Gráficos de puntos (dot-plot) de la población CD44+/CD24-/low (cuadrante Q1) en las células MCF-7/TC (izquierda) y MCF-7/CR (derecha). Se muestra la cuantificación de la población de células CD44+/CD24-/low en cada tipo celular. B. Gráficos de puntos (dot-plot) de la población CD133+ en las células MCF-7/TC (izquierda) y MCF-7/CR (derecha). Se muestra la cuantificación de la población de células CD133+ en cada tipo celular. Test Mann-Whitney. Se muestra la media ± EEM de 3 experimentos independientes.
Con el propósito de determinar si la sobreexpresión de HMGCR inducía la expresión de genes asociados a célula madre en la línea MCF-7, se procedió a evaluar marcadores de pluripotencia y de CMTs por RT-qPCR a las 48 horas post-transfección. Se analizaron los genes de pluripotencia clásicos de Yamanaka (Oct4, Sox2, Klf4 y cMyc), además de Nanog, encontrándose una tendencia a mayor expresión de Sox2, un aumento significativo de Nanog y una disminución significativa del oncogén Klf4 en las células MCF-7/CR. Sin embargo, no se encontraron diferencias significativas de expresión para Oct4 y cMyc. Por otro lado, se evaluaron marcadores de CMT en CM (BCRP, e- cadherina y vimentina), sin encontrarse diferencias entre ambos tipos celulares (Figura 8).
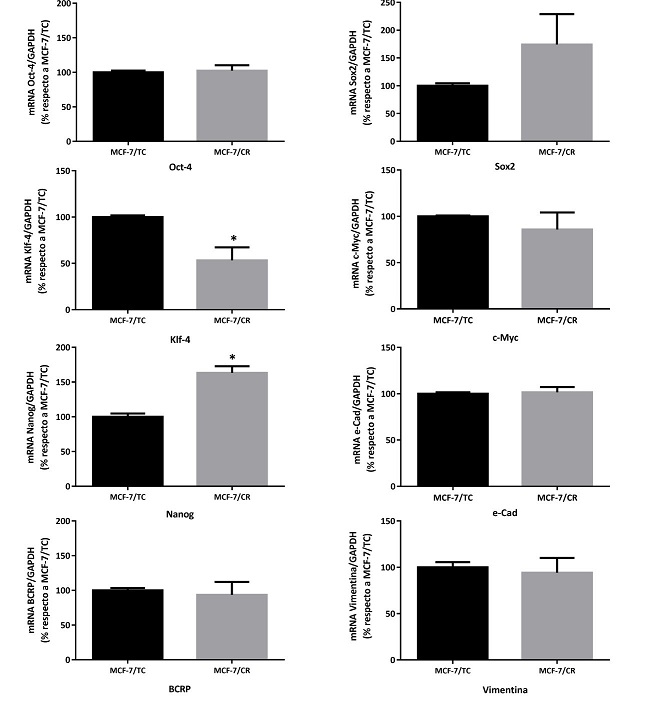
Figura 8: Caracterización molecular del sistema HMGCRon. Niveles de expresión de los marcadores Oct4, Sox2, Klf4, cMyc, Nanog, e-cad, BCRP y Vimentina por RT-qPCR. Test Mann- Whitney (*p<0.05 vs. MCF-7/TC). Se muestra la media ± EEM de 3 experimentos independientes.
> Propiedades celulares generales
Luego, se decidió seguir con la caracterización del modelo con inducción endógena de HMGCR, evaluando diversas propiedades relacionadas a la tumorigénesis. Con este fin, se realizaron ensayos de crecimiento de colonias en agar blando para evaluar la capacidad de crecer en forma independiente de anclaje, que representa una de las características distintivas de las células tumorales.
Si bien se obtuvieron colonias en ambos tipos celulares (MCF-7/TC y MCF-7/CR) y, a simple vista, no parecieron observarse diferencias entre ambas líneas en cuanto a morfología o tamaño de colonias, por problemas técnicos no se pudo realizar una cuantificación precisa. Por lo tanto, con estos ensayos no se logró determinar el efecto de la inducción de HMGCR sobre la capacidad de crecer sin estar adheridas a un sustrato (Figura 9.A).
Luego, se decidió estudiar el efecto que podría tener sobre la proliferación celular, sin encontrarse diferencias significativas entre ambos tipos celulares (Figura 9.B). Por último, se encontró que la línea MCF-7/CR presenta tendencia a menor adhesión (Figura 9.C) y concordantemente, a mayor migración (Figura 9.D) que la línea control, indicando que HMGCR podría estar involucrada en estos procesos.
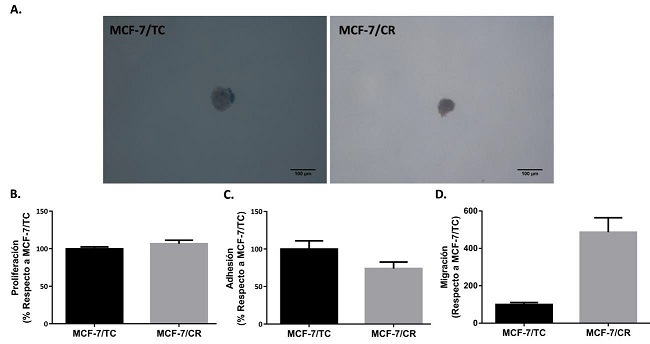
Figura 9: Caracterización de propiedades tumorales del sistema HMGCRon a 48 horas post- transfección. A. Imágenes representativas de colonias formadas en agar blando. B. Ensayo de proliferación (n=5). C. Ensayo de Adhesión (n=3). D. Ensayo de migración (n=2). Test Mann- Whitney. Se muestra la media ± EEM de experimentos independientes.
Continúa parte II en la siguiente página