La leucemia congénita (CL) se refiere a una leucemia que se presenta desde el nacimiento o se desarrolla en las primeras semanas de vida. Excluyendo la mieloproliferación neonatal transitoria relacionada con el síndrome de Down, la incidencia de CL es menor del 1% de las leucemias de la niñez. La leucemia cutis (LC) se caracteriza por infiltración de la piel por células hematopoyéticas malignas inmaduras, que se observan en el 30-50% de los pacientes con CL. Clínicamente, los pacientes pueden desarrollar un rash con nódulos eritematosos azulados de tamaño variado con una distribución generalizada. Rara vez están presentes otras lesiones que incluyen máculas, vesículo-pústulas y púrpura. Las mucosas generalmente no están afectadas. Los diagnósticos diferenciales incluyen infecciones congénitas incluyendo TORCH (toxoplasmosis, rubeola, citomegalovirus, herpes simple), sífilis congénita, histiocitosis de células de Langerhans, mastocitosis cutánea y enfermedades hematológicas.
La biopsia de piel se requiere para realizar el diagnóstico. El pronóstico de la leucemia congénita es generalmente grave, pero el diagnóstico y tratamiento temprano puede mejorar el pronóstico.
Se reporta el caso de un neonato con leucemia cutis congénita, confirmándose como leucemia mieloide aguda congénita (AML) del tipo M2 con defecto cromosómico t (8; 21) (p11; q22), y fue tratado exitosamente con quimioterapia para leucemia mieloide.
Reporte del caso:
Se presenta el caso de una niña de 19 días con antecedentes de un rash cutáneo asintomático rápidamente progresivo en el tronco, cara y miembros, presentes desde el nacimiento.
No presentaba síntomas sistémicos asociados. Nació a término de parto vaginal, con un peso al nacer de 3.2 kg. No presentaba historia familiar de enfermedad similar.
La madre negaba antecedentes de ingesta a drogas, infecciones, exposición a radiaciones y teratógenos durante el embarazo. El test TORCH fue negativo. Al exámen físico el peso, altura y circunferencia de cabeza correspondían al percentilo 50.
Presentaba múltiples pápulas y nódulos violáceos no dolorosos, firmes a gomoso en consistencia, en cara, cuero cabelludo y miembros (fig 1). El signo de Darier era positivo en una de las lesiones del tronco.
Presentaba distensión leve de abdomen con hepatoesplenomegalia (60 mm debajo del reborde costal). El resto del exámen físico fue normal.
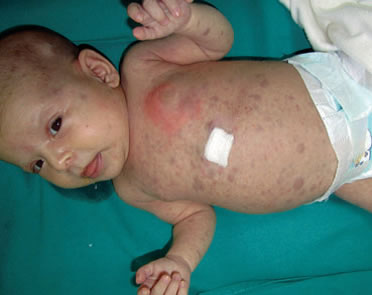
Figura 1. Erupción pápulo nodular violácea extensa.
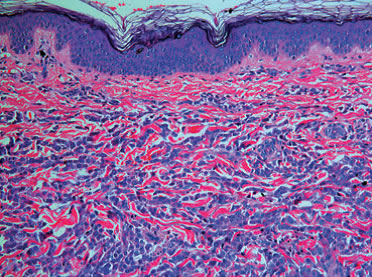
El exámen histológico de la biopsia de piel mostró una infiltración difusa de células leucémicas en dermis (fig 2 a) y tejido subcutáneo. Casi el 50% de las células eran positivas para CD15 (fig 2b), naphthol cloroacetato esterasa, antígeno específico mieloide (MSA) y CD117.
A
B
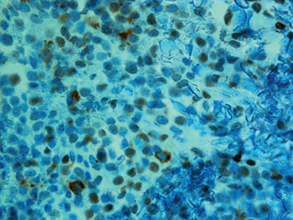
Figura 2 (a) Infiltación difusa de la dermis con células mieloides inmaduras (H&E). (b) Variaciones en la madurez del infiltrado dérmico de células blásticas que se observan en la tinción inmunohistoquímica con CD 15.
El hemograma completo mostró incremento de glóbulos blancos (21x109 ⁄ L; normal 4–11x109 ⁄ L) con 44% de linfocitos rango normal de 40-60%), y la presencia de células que normalmente no están presentes: 30% de células blásticas, 2% de metamielocitos y 6% de mielocitos. La paciente presentaba un bajo recuento de plaquetas 57 x 109 (rango normal 150-400 x 109) y hemoglobina normal (15.7 g/dL). Un recuento de glóbulos blancos luego de 1 semana mostró un incremento de glóbulos blancos (26.6 x 109) con 41% de células blásticas. El inmunofenotipo de sangre periférica mostró que las células blásticas eran positivas para CD 13, CD14, CD15, CD33, CD45, CD64, antígeno leucocitario humano (HLA)-DR y mieloperoxidasa.
La tinción del aspirado de medula ósea con Sudan B mostró más del 30% de células blásticas conteniendo gránulos (fig 3). Se diagnosticó leucemia mieloide aguda (AML), con una clasificación de AML-M2 basada en las características morfológicas y los resultados de las tinciones especiales. En el análisis citogenético del aspirado de médula ósea, se observó un defecto cromosómico, cariotipo 46, XX con t (8;21) (p11; q 22).
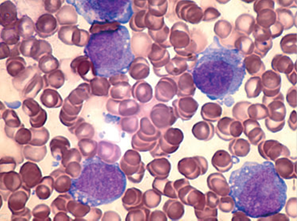
Figura 3 Células rojas y blastos con citoplasma granular (mieloblastos) y
nucleolos prominentes en aspirado de medula ósea.(Sudan B).
La paciente recibió 3 cursos de quimioterapia (citarabina, daunorrubicina y etopósido) a intervalos de 3 meses. Las lesiones desaparecieron rápidamente luego del primer curso de quimioterapia, y no hubo recurrencia de las lesiones cutáneas durante el seguimiento.
En una revisión reciente de LC en 80 neonatos, la mayoría de los casos estaban asociados a AML, siendo el principal subtipo la leucemia monocítica M5. La aparición de las lesiones cutáneas y el diagnóstico de leucemia ocurre al mismo tiempo en el 85% de los pacientes. El diagnóstico de leucemia luego de los nódulos cutáneos (1 semana a 3 meses) se realizó en 12.5% de los pacientes, y precedió las lesiones cutáneas (1.8 semanas) en el 2.5%. La apariencia típica estaba presente en el 85% de los casos. Rara vez se han reportado casos de LC aleucémica, presentando lesiones cutáneas leucémicas sin enfermedad sistémica ni compromiso de la médula ósea.
Se ha reportado recientemente una ocurrencia familiar. La paciente presentada tenía AML M2, y el diagnóstico de AML se efectuó cuando presentó el rash cutáneo. La patogénesis de LC permanece desconocida. Se ha propuesto que varios factores de riesgo maternos se asocian con el desarrollo de CL, incluyendo el consumo de alcohol, tabaco e inhibidores de la topoisomerasa II (de plantas flavonoides) por la madre, y su exposición a radiación y altos niveles de factores de crecimiento símil insulina. Esos factores de riesgo no estaban presentes en la madre de la paciente.
La asociación de CL con anormalidades cromosómicas condujo a especular si la CL es resultado de una fragilidad cromosómica.
Las delecciones cromosómicas son eventos moleculares comunes en malignidades mieloides, y estas delecciones se propone que contienen genes de supresión tumoral que ocasionan síndromes mielodisplásicos.
Como la hematopoyesis embriogénica comienza en el mesénquima indiferenciado, que comienza en la tercera semana luego de la fertilización, LC puede ser un evento primario y la primera presentación de CL..
Se ha reportado un porcentaje de sobrevida del 37.5% en neonatos con LC. Si no se trata, generalmente es fatal.
Sin embargo, en casos aislados se logró remisión espontánea temporaria o permanente sin intervención de quimioterapia. En esos casos es controversial la decisión de tratar o no tratar. Algunos autores recomiendan que en ausencia de progresión de la enfermedad o traslocación 11q23, la CL puede manejarse en forma conservadora esperando una remisión espontánea. Los autores decidieron tratar a la paciente con quimioterapia por la extensión de las lesiones y el curso rápidamente progresivo. Presentó una respuesta favorable a la quimioterapia y continuaba bien a los 9 meses de seguimiento.
La LC congénita es una presentación rara de leucemia congénita que aparece al nacer o en el periodo neonatal.
Clínicamente, se caracteriza por una erupción generalizada pápulo-nodular violácea.
Histológicamente, se caracteriza por la infiltración de la piel (dermis y tejido subcutáneo) por células mieloides inmaduras.
El rash precede, sigue o se presenta simultáneamente con el inicio de la leucemia.
Si no se trata, el pronóstico es generalmente pobre, pero el diagnóstico y tratamiento temprano producen un mejor resultado.
¿Qué aporta este artículo a la práctica dermatológica?.
La leucemia congénita (CL) es una malignidad rara que representa menos del 1% de los casos de leucemias de la niñez. La leucemia cutis (LC) se refiere a una infiltración cutánea con células leucémicas, y se observa en el 30-50% de los casos de CL. Puede preceder, seguir u ocurrir simultáneamente con leucemia. Si no se trata el pronóstico es pobre, pero el diagnóstico temprano y el tratamiento puede resultar en un pronóstico favorable. Se reporta el caso de una leucemia cutis congénita con una erupción pápulo nodular violácea progresiva, presente desde el nacimiento, como un signo de presentación de leucemia mieloide aguda (AML), que se clasificó como tipo AML.FAB M2 con un defecto cromosómico t (8;21) (p11;q22). La paciente presentó una respuesta favorable a la quimioterapia AML.
♦ Comentario y resúmen objetivo: Dra. Geraldina Rodriguez Rivello.