El síndrome de anticuerpos antifosfolípidos (APS) puede causar eventos trombóticos catastróficos, con manifestaciones cutáneas como primer signo de APS en casi un 40% de los pacientes. Es importante diagnosticarlo apropiadamente y comenzar el tratamiento con anticoagulación para evitar el daño irreversible secundario a la trombosis.
Se presenta el caso de una mujer de 51 años con antecedentes de una úlcera crónica en la extremidad inferior izquierda. La úlcera parecía infectada por lo que se trató con cefalexina durante 1 semana. No se observó mejoría con cefalexina por lo que se suspendió y se inició trimetroprima/sulfametoxazol. Al tercer día del tratamiento con trimetoprima/sulfametoxazol desarrolló múltiples lesiones en las extremidades inferiores asociadas a fiebre y mal estado general. Las lesiones progresaron en forma difusa por todo el cuerpo en los 4 días siguientes. Al día 7 de tratamiento con trimetoprima/sulfametoxazol, notó sus tobillos purpúricos y negros. Presentaba antecedentes de madre con stroke a los 40 años e infarto de miocardio fatal a los 50 años y dos primos con trastornos inespecíficos de la coagulación.
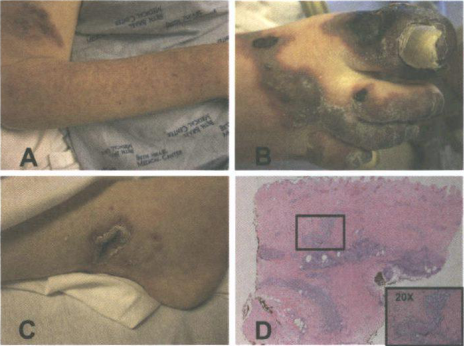
Figura 1: Hallazgos clínicos e histopatología. A. Numerosas máculas eritematosas menores de 1mm, ubicadas en extremidades. B. Múltiples placas purpúricas con bordes eritematosas, zonas negras, áreas necróticas y ampollas flácidas. C. Ulcera seca de 2 cm con bordes eritematosos indurados y cicatriz blanca. D. Con la tinción de hematoxilina y eosina se revela una trombosis difusa con un infiltrado celular mixto intramural y perivascular y ausencia de vasculitis.
Al exámen físico la paciente se presentaba afebril, con numerosas máculas eritematosas menores de 1 mm ubicadas en extremidades (figura 1 A). En ambos pies presentaba placas purpúricas múltiples con bordes eritematosos. Algunas áreas eran negras y necróticas y otras presentaban ampollas flácidas (figura 2 B). Las áreas afectadas eran frías a la palpación. En la parte medial de la extremidad inferior izquierda presentaba una úlcera seca de 2 cm con bordes indurados eritematosos y una cicatriz blanca (figura 1 C), los pulsos pedios, perineales y poplíteos eran 3+.
La determinación inicial de laboratorio reveló anemia con esquistocitos y esferocitos, trombocitopenia, coagulopatía con tests de función hepática, lactato deshidrogenasa, eritrosedimentación (ESR), proteina C reactiva (PCR), inmunoglobulina IgM e IgG séricas elevados. Presentaba un test anticoagulante lúpico positivo y anticuerpo IgM B2 microglobulina positivo.
La histopatología reveló trombosis difusa con un infiltrado celular mixto intramural y perivascular y ausencia de vasculitis (figura 1D). Todos los cultivos bacterianos eran negativos y los estudios vasculares normales.
Los resultados eran compatibles con APS según el consenso internacional. Apoyaban el diagnóstico; a) la edad relativamente jóven, b) antecedentes maternos de accidente cerebrovascular y ataque cardíaco, c) gatillado por trimetoprima/sulfametoxazol, d) afebril sin dolor articular ni otros síntomas sistémicos, e) la no cicatricación de la úlcera de extremidad inferior de 2 años de evolución a pesar de pulsos periféricos fuertes, hemoglobina A1c dentro de límites normales sin otras secuelas de enfermedad vascular, f) presentación clínica de púrpura retiforme y necrosis digital, g) hallazgos de laboratorio, incluyendo niveles elevados de ESR, PCR, IgG, IgM, trombocitopenia y anemia hemolítica.
La paciente comenzó con prednisona 60 mg día y en los próximos 7 días, el rash petequial difuso mejoró, pero las placas purpúricas en dedos de pies y pies empeoraron progresivamente. Una vez confirmado el diagnóstico de APS, se inició anticoagulación con enoxiheparina y warfarina, tratamientos considerados de primera elección en APS. Los corticoides, inmunoglobulina intravenosa y ciclofosfamida pueden ser beneficiosos, pero no existen estudios largos controlados por lo que se consideran tratamientos de segunda línea para casos refractarios.
A los 6 meses de seguimiento las lesiones curaron completamente con mínima pérdida del pulpejo del primer dedo de pie derecho. La paciente se encuentra actualmente con profilaxis a largo plazo con warfarina sin recurrencia de la enfermedad.
¿Qué aporta éste artículo a la práctica dermatológica?
El síndrome de anticuerpos antifosfolípidos se produce por autoanticuerpos de la superficie celular de fosfolípidos o de proteínas de unión de fosfolípidos ocasionando anormalidades de la coagulación con secuelas devastadoras, incluyendo accidente cerebrovascular, trombosis venosa profunda, embolismo pulmonar y abortos espontáneos recurrentes. Sin embargo, las manifestaciones cutáneas representan el primer signo de APS en casi el 41% de los pacientes.
♦ Comentario y resúmen objetivo: Dra. Geraldina Rodríguez Rivello.