Los síndromes paraneoplásicos son un grupo de enfermedades asociadas a malignidad. Reflejan comunicaciones patológicas entre las células tumorales y las células del huésped en un sitio remoto del tumor primario y de sus metástasis. Una lesión dermatológica se considera paraneoplásica si ocurre en alto porcentaje en asociación a malignidad en relación a la población general; aparece antes, al mismo tiempo, o posterior al diagnóstico de malignidad, y mejora luego del tratamiento exitoso de la malignidad.
La pitiriasis rotunda (PR) fue inicialmente descripta por Toyoma en 1906, quien la denominó pitiriasis circinada.
Esta condición ocurre más comúnmente en poblaciones japonesas, oeste de India y Sudáfrica, y en el último caso ocurre más comúnmente en poblaciones negras. Se ha descripto en nativos de Sardinia. Se ha reportado un número de casos de PR en poblaciones blancas.
En Ciskei, Sud Africa, la prevalencia de PR fue de 63 de 5800 (1.1%) en pacientes negros de un hospital rural. La edad media de estos pacientes fue de 60 años (rango 18-89 años), que fue más alta que la edad típica reportada en la literatura.
En la búsqueda de PubMed, se encontraron dos reportes de casos de mieloma múltiple asociado con PR. El primer paciente fue diagnosticado en 1974 con mieloma múltiple IgG y cirrosis, y el otro se diagnosticó en 1991 con mieloma múltiple.
Se presentan dos pacientes con diagnóstico de mieloma múltiple y PR. El diagnóstico de mieloma múltiple se confirmó acorde a los criterios propuestos por el grupo de trabajo internacional de mieloma como se muestra en la tabla 1.
Reporte
Paciente 1.
Mujer de 68 años, raza negra de Bloemfontein, con diagnóstico de mieloma múltiple en el 2009, luego de una fractura de cadera patológica con plasmocitoma de fémur, confirmado por histología. La paciente se perdió en el seguimiento. Consultó a los 2 años con deterioro general y progresión del mieloma múltiple. No presentaba antecedentes familiares de enfermedades dermatológicas, HIV negativo. Al exámen dermatológico, la paciente presentaba lesiones redondas, escamosas distribuídas en glúteos, tórax y parte superior de muslos (Fig 1). Las lesiones eran hiperpigmentadas, con bordes bien delimitados, entre 40 y 60 mm de diámetro, asintomáticas.
La paciente no pudo especificar la duración de las lesiones. No tenía antecedentes de recibir cremas con corticoides. Se confirmó el diagnóstico de PR.
Al exámen dermatológico, no se encontró ictiosis. Se excluyó el carcinoma hepatocelular por el hallazgo normal del nivel de a fetoproteína (5.1 g/L; rango de referencia 0.0-7.0 g/L) y ecografía abdominal normal.
La paciente presentaba hipertensión, y se le diagnosticó una cardiopatía dilatada, con una fracción de eyección ventricular izquierda de 21% por ecocardiograma, también presentaba hipotiroidismo TSH 17.8 mU/L (rango de referencia 0.27-4.2 mU/mL) y triiodotironina 1.7 lg/dL (12.0-22.0 lg/dL).
Manifestó angioedema agudo el segundo día de internación, y reportó ataques similares los 5 años previos. Recibía perindopril para hipertensión previo a la admisión; en la internación se rotó a enalapril. Los niveles de inhibidor esterasa C1 y C4 eran normales durante el episodio agudo.
Paciente 2.
Mujer de 60 años, negra de sud Africa, que presentaba, dolor de espalda crónico y se diagnosticó mieloma múltiple en marzo del 2012. Aunque presentaba colapso de la cuarta y quinta vértebra lumbar, no presentaba dolores neurológicos ni síntomas además del dolor.
La paciente no presentaba hallazgos de amiloidosis sistémica y era HIV negativa. El exámen físico reveló tres lesiones grandes circulares en el tronco del paciente. La de mayor tamaño estaba presente en el abdomen (Fig 1). Las lesiones eran hiperpigmentadas, bien delimitadas, cubiertas de una escama fina. Asintomáticas. No presentaba ictiosis. Se confirmó PR por la apariencia clínica, la confirmación histológica no se consideró necesaria.
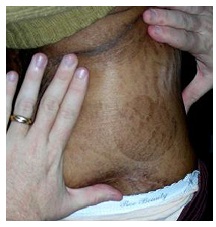
Figura 1: Lesión redonda, escamosa de pitiriasis rotunda en una mujer con inmunodeficiencia y mieloma múltiple.
No presentó una internación complicada, y se inició quimioterapia combinada CVAD (ciclofosfamida, vincristina, adriamicina y dexametasona) para el manejo del mieloma múltiple. Lamentablemente, ambas pacientes se perdieron en el seguimiento.
La PR es una dermatosis rara, idiopática, crónica, caracterizada por lesiones pigmentadas, redondas, bien delimitadas, escamosas predominantemente en tronco, pero también en espalda, glúteos y extremidades proximales. Existe una distribución igual por sexo, y típicamente ocurre entre los 20 y 45 años de edad. Las lesiones de PR pueden ser redondas u ovales, y pueden variar en tamaño y número, son generalmente asintomáticas.
Las lesiones parecen hipopigmentadas en pieles claras e hiperpigmentadas en pieles oscuras. El diagnóstico se hace generalmente por la apariencia clínica, y se observan hallazgos similares a la ictiosis vulgar en el estudio histológico.
Se han propuesto dos tipos de PR. El tipo 1 generalmente en personas negras, es no familiar e hiperpigmentada. Se ha identificado una asociación con infecciones sistémicas o malignidades. El tipo 2 generalmente ocurre en personas blancas como parches hipopigmentados, con antecedentes familiares positivos y condición subyacente no identificable.
En 1989 un estudio del hospital Baragwanath, South Africa, confirmó una relación estadísticamente significativa entre el carcinoma hepatocelular y PR en poblaciones negras de sud Africa.
La tabla 2, resúme las asociaciones de la enfermedad en 63 pacientes con PR publicados por Swift y Saxe. Estos autores concluyen que la mayoría de los casos se asociaron con tuberculosis (36.5%) y enfermedad hepática (22.2%), y podrían considerarse como marcadores de enfermedad seria.
En las dos pacientes, las características clínicas de PR fueron compatibles con los hallazgos previamente descriptos en la literatura en pacientes con mieloma múltiple.
Las gammapatías monoclonales, incluyendo el mieloma múltiple, se han asociado con varias enfermedades dermatológicas. La PR corresponde a un grupo de enfermedades cutáneas misceláneas descriptas en asociación con gammapatías monoclonales.
Según el conocimiento de los autores, se han reportado previamente solo 2 casos de PR en pacientes con mieloma múltiple.
Los autores reportan dos casos de PR detectados como hallazgos paraneoplásicos incidentales en pacientes femeninas negras con mieloma múltiple. El hallazgo de PR debería alertar a los médicos sobre la posibilidad de malignidad subyacente, incluyendo las discrasias de células plasmáticas.
La PR se asocia con condiciones serias, incluyendo malignidades. Aunque la PR puede ocurrir en pacientes con mieloma múltiple, esta asociación se ha reportado raramente. Se describen dos casos de PR como fenómeno paraneoplásico en pacientes con mieloma múltiple, destacando esta rara pero clínicamente importante asociación.
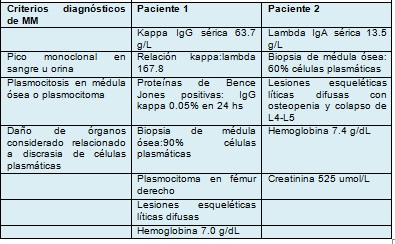
Tabla 1 Criterios diagnósticos de mieloma múltiple (MM) y hallazgos de dos pacientes con pitiriasis rotunda concurrente. 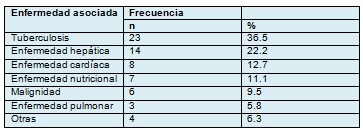
Tabla 2 . Enfermedades asociadas con pitiriasis rotunda en 63 pacientes.
¿Qué aporta este artículo a la práctica dermatológica?.
La pitiriasis rotunda (PR) es una dermatosis crónica poco común, que puede ser idiopática o puede asociarse con infecciones y malignidad. Se describen los hallazgos clínicos y bioquímicos en dos pacientes con mieloma múltiple, a los que se les diagnosticó PR, y se detallan las condiciones clínicas con las que puede asociarse este raro fenómeno paraneoplásico. Aunque la PR es una condición rara, se puede encontrar como un fenómeno paraneoplásico en varias condiciones, y por lo tanto debería reconocerse como un signo clínico importante.
Comentario y resúmen objetivo: Dra. Geraldina Rodríguez Rivello