| Aspectos destacados |
|
• La linfocitosis es comúnmente causada por infecciones virales transitorias, pero puede persistir en infecciones crónicas (hepatitis B, hepatitis C, VIH o tuberculosis) y leucemia linfocítica crónica (LLC). • Siempre se debe controlar e investigar la linfocitosis, incluida la realización de un hemograma completo con fórmula leucocitaria, frotis de sangre y examen de los ganglios linfáticos y el abdomen. • La leucemia linfocítica crónica (LLC) se diagnostica cuando hay ≥5.0 × 109/L linfocitos B monoclonales (confirmados mediante inmunofenotipado por citometría de flujo) y un frotis sanguíneo que confirma linfocitosis, células leucémicas pequeñas y de apariencia madura. • Se adopta una estrategia de observación cuando se trata a pacientes con LLC con enfermedad asintomática no activa. Cuando los pacientes tienen una enfermedad sintomática activa, se recomienda el tratamiento con quimioinmunoterapia. • La identificación de mutaciones genéticas es importante para guiar el tratamiento óptimo y ayuda a informar el pronóstico. |
| Introducción |
La linfocitosis se define como un recuento de linfocitos ≥5.0 x 109/L y comúnmente es causada por infecciones virales transitorias.
La linfocitosis también se puede observar en enfermedades crónicas (hepatitis B, hepatitis C, VIH y tuberculosis) y en la leucemia linfocítica crónica (LLC). Por lo tanto, una linfocitosis siempre debe ser monitoreada e investigada (Figura 1). Cuando el recuento de linfocitos se eleva de forma persistente, se requiere el aporte de un especialista en hematología.
La leucemia linfocítica crónica (LLC) es la leucemia más común en el mundo occidental, con una mediana de edad de diagnóstico de 70 años. La LLC implica una expansión monoclonal maligna de linfocitos B, con infiltración progresiva en los ganglios linfáticos y sitios de hematopoyesis, incluidos el hígado, el bazo y la médula ósea. Como tal, los pacientes pueden presentar linfadenopatía, hepatoesplenomegalia y citopenia dependiendo del grado de progresión de la enfermedad.
Morfológicamente, los linfocitos en la leucemia linfocítica crónica (LLC) parecen pequeños y maduros, pero generalmente no reaccionan y son disfuncionales, lo que resulta en susceptibilidad a infecciones causadas por hipogammaglobulinemia. Es inusual que los pacientes con LLC presenten los clásicos "síntomas B", como sudores nocturnos, fiebre y pérdida de peso. De hecho, la mayoría de los diagnósticos se identifican debido a un hallazgo incidental de linfocitosis en análisis de sangre de rutina.
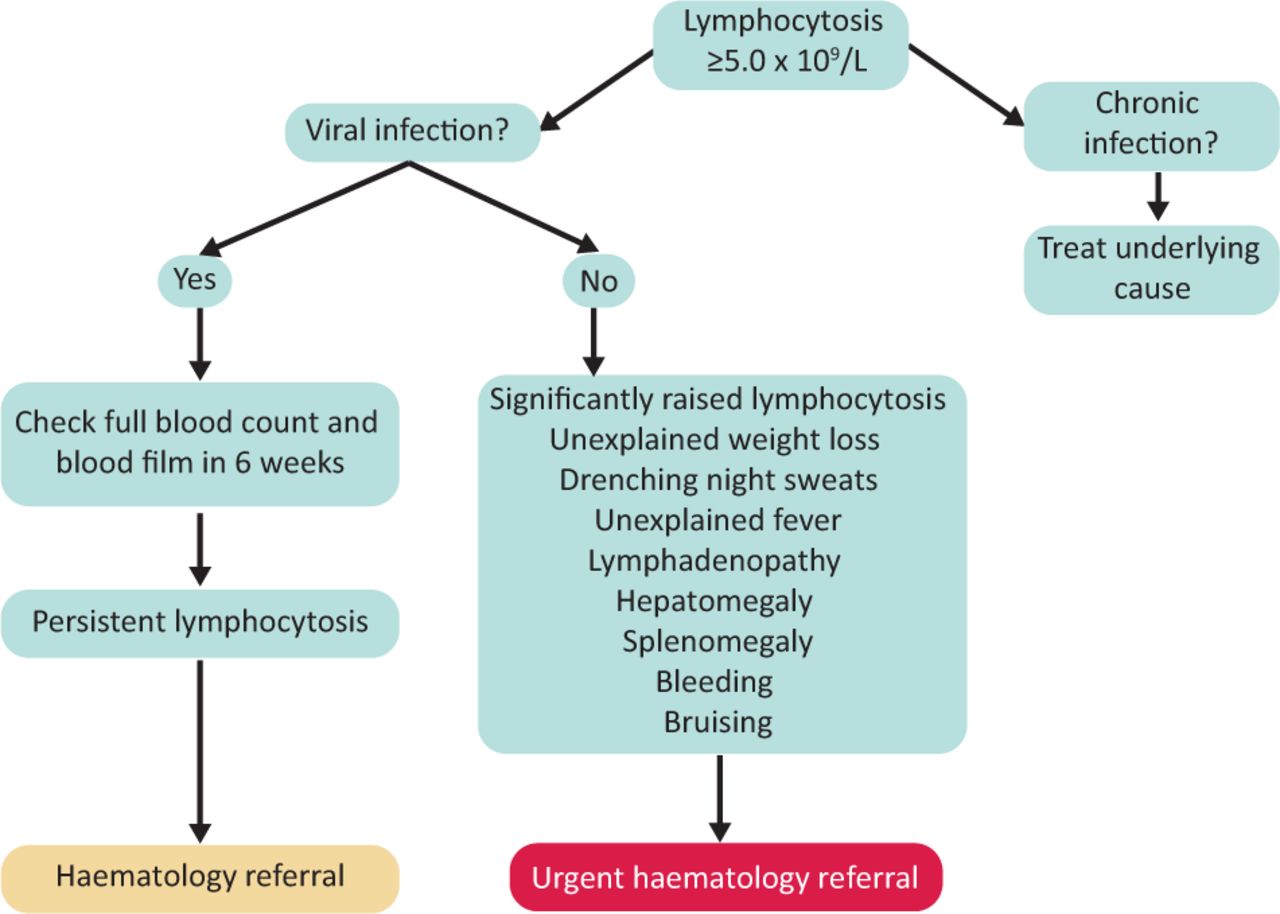
Figura 1. Vía de derivación de un paciente con linfocitosis.
| Examen |
Se debe realizar un examen completo de los ganglios linfáticos cervicales, axilares e inguinales, así como un examen abdominal en busca de signos de hepatoesplenomegalia, que se observa en aproximadamente el 50% de los casos.
Los signos de insuficiencia de la médula ósea pueden manifestarse como petequias o palidez. En el caso de que un paciente tenga alguno de los signos mencionados, con linfocitosis confirmada y sin infección viral reciente, se requiere una derivación urgente a hematología (Figura 1).
| Investigaciones |
La primera investigación requerida en caso de sospecha de leucemia linfocítica crónica (LLC) es un recuento sanguíneo completo, con un valor de glóbulos blancos que muestra linfocitos ≥5.0 × 109/L, indicando la necesidad de un frotis de sangre para evaluar la morfología y confirmar la linfocitosis. Se realizará una citometría de flujo para confirmar la clonalidad de los linfocitos B circulantes.
Los marcadores inmunofenotípicos característicos de la LLC incluyen CD5, CD19, CD20 y CD23. Las células son inmaduras y frágiles y se dañan durante la preparación del portaobjetos, dando así las características células de mancha que se ven al frotis. Juntos, la presencia de ≥5.0 x 109/L linfocitos B monoclonales y estos hallazgos característicos al microscopio, establecen el diagnóstico de LLC.
El análisis citogenético mediante fluorescencia es necesario para detectar la deleción del cromosoma 17 (del17p), que contiene el gen TP53 en su brazo corto, así como la secuenciación de genes para identificar mutaciones codificantes. Este es un gen supresor de tumores, a menudo denominado "guardián del genoma", y su eliminación o mutación se asocia con tasas de respuesta al tratamiento más bajas y una supervivencia libre de progresión más corta. La identificación de del17p o la presencia de mutación TP53 es clave para guiar los regímenes de tratamiento y ayuda a informar el pronóstico.
La lactato deshidrogenasa (LDH) y el test de antiglobulina directa (TAD) son investigaciones prácticas en pacientes anémicos y pueden ser útiles para identificar aquellos con anemia hemolítica relacionada con autoinmunidad (AHAI) que puede complicar la leucemia linfocítica crónica (LLC) y debe impulsar una revisión clínica urgente.
La púrpura trombocitopénica inmune (PTI) es otra complicación autoinmune de la leucemia linfocítica crónica (LLC) que puede causar plaquetopenia considerable.
En pacientes con infecciones recurrentes, se deben controlar los niveles de inmunoglobulina para detectar hipogammaglobulinemia. Se debe establecer el estado de hepatitis B, hepatitis C, citomegalovirus y VIH antes de cualquier quimioterapia-inmunoterapia para prevenir la reactivación del virus.
Aunque las biopsias por aspiración de médula ósea no son necesarias para un diagnóstico formal de leucemia linfocítica crónica (LLC), pueden ser útiles cuando el diagnóstico no está claro y para determinar la respuesta al tratamiento si es necesario.
La tomografía computarizada (TC) del cuello, el tórax, el abdomen y la pelvis a menudo se realiza en el estudio previo al tratamiento de quimioinmunoterapia. Las imágenes pueden evaluar la carga tumoral y establecer el riesgo de síndrome de lisis tumoral.
Donde hay ganglios linfáticos que crecen rápidamente, la biopsia ganglionar tiene un papel en el establecimiento de una posible transformación de la leucemia linfocítica crónica (LLC) en un linfoma de células B de alto grado. Este proceso se conoce como "transformación de Richter", y los pacientes suelen informar síntomas de fiebre y pérdida de peso.
| Manejo |
La quimioinmunoterapia es la principal forma de tratamiento de la leucemia linfocítica crónica (LLC) y puede prolongar la supervivencia.
A pesar de esto, no a todos los pacientes se les ofrece, ya que la terapéutica de la enfermedad en etapa temprana no ha demostrado evidencia de beneficio. Se adopta una estrategia de observación y espera cuando se trata a estos pacientes, que incluye hemogramas regulares cada 3 a 12 meses, dependiendo del tiempo de duplicación de linfocitos, y exámenes físicos completos para evaluar la progresión de su LLC.
Cuando los pacientes tienen una enfermedad sintomática activa, hay opciones de quimioinmunoterapia disponibles. Los ejemplos de enfermedad activa consisten en una pérdida de peso >10%, fatiga significativa, sudores nocturnos, evidencia de insuficiencia medular progresiva (anemia y trombocitopenia), un aumento en el recuento de linfocitos ≥50% en 2 meses o tiempo de duplicación rápido, hepatoesplenomegalia progresiva y linfadenopatía. Deben ser derivados urgentemente a hematología para iniciar el tratamiento (Figura 1).
La quimioinmunoterapia se dirige a las células cancerosas que se dividen rápidamente, pero es importante tener en cuenta que los sitios de alto recambio celular (por ejemplo, el revestimiento de la mucosa oral, el revestimiento de la mucosa digestiva, las palmas de las manos, la parte plantar de los pies y la médula ósea) también se ven afectados y dan como resultado efectos secundarios significativos. Estos incluyen úlceras orales, náuseas y vómitos, disestesia palmoplantar, hematomas, sangrado e infecciones secundarias a citopenias.
En vista de estos efectos secundarios, se debe considerar el estado funcional de los pacientes para determinar si están lo suficientemente en forma para tolerar el tratamiento. A los que inician quimioinmunoterapia se les prescriben antibióticos profilácticos, aciclovir, alopurinol, colutorio de clorhexidina y antieméticos para mitigar los efectos secundarios. Cuando los pacientes tengan trombocitopenia o anemia, se deben proporcionar hemocomponentes, según sea necesario.
| Regímenes de quimioinmunoterapia |
La presencia de del 17p o mutación TP53 guía los regímenes de quimioinmunoterapia. Cuando no existen tales mutaciones, se recomienda fludarabina, ciclofosfamida y rituximab (FCR) como tratamiento de primera línea. Cuando esto no sea adecuado, se pueden considerar bendamustina y rituximab (BR) o clorambucilo y obinutuzumab (CO). La ciclofosfamida, el clorambucilo y la bendamustina son ejemplos de agentes alquilantes que actúan directamente sobre el ADN para prevenir la división celular y reducir la masa total de linfocitos.
La fludarabina es un ejemplo de un análogo de purina que inhibe la síntesis de ADN. Puede causar linfopenia grave y provocar enfermedad de injerto versus huésped asociada a transfusiones; por tanto, los pacientes que reciben transfusiones de sangre necesitan productos sanguíneos irradiados.
Rituximab y obinutuzumab son anticuerpos monoclonales anti-CD20 quiméricos dirigidos a células B patógenas y normales. Más del 10% de los pacientes experimentan efectos secundarios relacionados con la infusión de rituximab, incluidos síntomas similares a los de la gripe y erupciones cutáneas, que también se observan con obinutuzumab, un anticuerpo más potente que puede inducir el síndrome de lisis tumoral.
| Nuevas terapias dirigidas para la LLC |
En presencia de del17p o mutación TP53, se recomienda el tratamiento con ibrutinib (o acalabrutinib) o venetoclax, con o sin obinutuzumab (VO) o rituximab (VR). VO y acalabrutinib también están autorizados en el entorno de primera línea donde FCR, BR o clorambucilo se consideran inadecuados. Ibrutinib y acalabrutinib actúan inhibiendo la tirosina quinasa de Bruton, que es un componente clave en la señalización de supervivencia de las células receptoras de células B.
| Estudio de un caso |
|
Presentación Un hombre de 51 años fue revisado por su médico general ya que informó dolor abdominal generalizado y antecedentes de sudores nocturnos durante las últimas 6 semanas. Negó pérdida de peso y refería estar bien y en forma. En el examen físico, tenía un ganglio linfático cervical de 2 cm y un ganglio linfático axilar de 2 cm en el lado izquierdo. No presentaba hepatomegalia ni esplenomegalia. Su médico de cabecera realizó algunos análisis de sangre de referencia y se encontró que el paciente tenía un recuento de linfocitos de 70,8.×109/L Se realizó derivación urgente a hematología. Diagnóstico El paciente fue evaluado por el equipo de hematología. Para confirmar el diagnóstico se solicitó hemograma completo, frotis sanguíneo e inmunofenotipado. Otras pruebas incluyeron urea y electrolitos, pruebas de función hepática, prueba directa de antiglobulina, calcio, inmunoglobulinas y electroforesis de proteínas séricas. El paciente estaba anémico con hemoglobina de 93 g/L y tenía una linfocitosis de 72 x 109/L con fragmentos de células en el frotis. La citometría de flujo confirmó los linfocitos B positivos para CD5 y CD23. Se le realizó un aspirado de médula ósea que mostró un infiltrado linfocitario del 86%. Se le diagnosticó LLC basándose en un inmunofenotipo confirmatorio. Tratamiento El paciente tenía un estado funcional de 0 y se preparó para iniciar quimioinmunoterapia. La citogenética no mostró del17p o mutación TP53 y se inició tratamiento con fludarabina, ciclofosfamida y rituximab (FCR) con antibióticos profilácticos y alopurinol. 5 años después su enfermedad recayó y se inició tratamiento con ibrutinib con buena respuesta. Venetoclax sería el tratamiento de elección en caso de requerirse tratamiento de tercera línea en el futuro. |
| Conclusiones |
Identificar la causa subyacente de una linfocitosis es fundamental en el manejo adecuado de los pacientes. Cuando el recuento de linfocitos se eleva de manera persistente o significativa y se han excluido causas benignas (infecciones virales o afecciones crónicas), se requiere la derivación inmediata a un hematólogo.
Se realiza un diagnóstico de leucemia linfocítica crónica (LLC) cuando hay ≥5.0 × 109/L linfocitos B monoclonales (basado en citometría de flujo confirmatoria) y un frotis de sangre de apoyo que muestra linfocitos maduros y células difuminadas. Investigaciones adicionales que incluyen citogenética, LDH, TAD, microglobulina B2, aspirado de médula ósea, detección viral y TC tienen un papel en la estadificación y determinación del pronóstico.
Los médicos de atención primaria desempeñan un papel clave en la identificación de pacientes con LLC con enfermedad activa y sintomática. La derivación a un especialista es imprescindible para evaluar la progresión de la enfermedad y las opciones de tratamiento disponibles.
Traducción, resumen y comentario objetivo: Dr. Esteban Crossio