El complemento es un elemento esencial del sistema inmune innato, y la ausencia de componentes del complemento puede causar severas alteraciones en la defensa del huésped. Se ha reportado que las deficiencias del componente C4 se asocian con lupus eritematoso sistémico (SLE), nefropatía por IgA, púrpura de Scholein-Henoch (HSP) y otras enfermedades del sistema inmune. En este reporte, se presenta el caso de una vasculitis cutánea y glomerulonefritis asociada con deficiencia de C4.
Reporte:
Se presenta el caso de una mujer de 44 años con un rash en sus piernas. Cinco meses previos, presentó resfrío, con dolor de garganta, dolores articulares y fiebre baja, seguida de edema leve y púrpura de miembros inferiores. Los síntomas coincidieron ocasionalmente con hematuria microscópica y proteínas en orina, controlada con furosemida y con un antiinflamatorio no esteroideo (loxoprofen). No presentaba antecedentes personales ni familiares relevantes.
Al examen físico, presentaba un rash compuesto por lesiones purpúricas palpables en ambas áreas pretibiales (Fig 1a). La paciente era sana. Debido a que los síntomas eran recalcitrantes, se tomaron biopsias de piel y renales.
Al examen histológico de una de las lesiones purpúricas, presentaba infiltración leve de neutrófilos con leucocitosis y extravasación de fibrina en dermis superior y media, hallazgos consistentes con vasculitis neutrofílica leucocitoclásica (Fig 1 b, c). La inmunofluorescencia directa reveló depósitos granulares de IgG, IgM, C3 y C1q en las paredes de los vasos en dermis superior. En la biopsia renal se observó glomerulonefritis proliferativa mesangial leve, con leve daño en los capilares (Fig 2 a, b). La inmunofluorescencia reveló depósitos granulares de Ig M y C3, principalmente en el mesangio pero también en la membrana basal de los capilares con una distribución segmentaria (Fig 2 c,d).
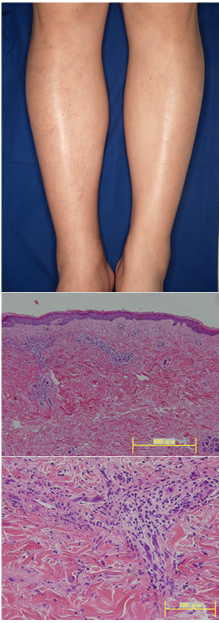
Figura 1. (a) Púrpura palpable en ambas pretibias; (b,c) biopsia de piel infiltración leve de neutrófilos con leucocitoclasia y extravasación de fibrina en dermis superior y media.
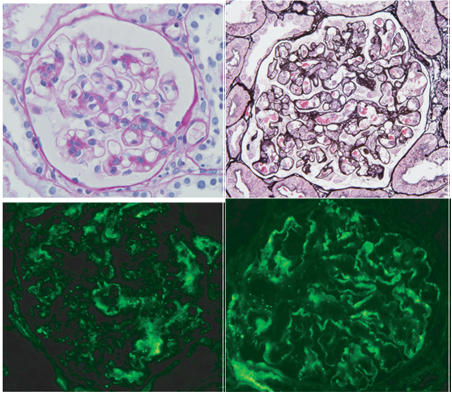
Figura 2. (a,b) Biopsia renal que muestra proliferación mesangial y engrosamiento de las paredes capilares en la membrane basal glomerular. (a) Acido periódico de Schiff; (b) y metenamida plata;(c,d) Inmunofluorescencia directa detectó depósitos granulares de (c) IgM y (d) C3, principalmente en el mesangio. Aumento (c,d) x 400.
Las investigaciones de laboratorio hemograma completo, tests de función renal y hepática, coagulación y fibrinólisis fueron normales.
El nivel de factor reumatoideo fue elevado (1083 UI/mL), rango normal menor de 10 UI/mL), IgM (342 mg/dL, 35-220) y aglutinación de partículas de artritis reumatoidea (x 640; menor de x 40), y bajos niveles de IgA (101 mg/dL, 120-510 mg/dL). Los tests fueron negativos para anticuerpos antinucleares, anticuerpo nuclear extraíble, y mieloperoxidasa y anticuerpos anticitoplasmáticos antineutrófilos proteinasa 3.
El nivel de C3 fue casi normal (79 mg/dL, 80-150/dL), pero el C4 era indectable (menor de 1.5 mg/dL, 15-40 mg/dL), la actividad hemolítica CH50 estaba reducida (12.1 U/ mL; 32-44 U/mL). El inhibidor esterasa C1 fue normal en el 99%.
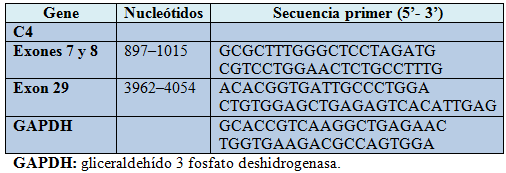
El análisis urinario reveló hematuria microscópica y proteínas en orina. Se comparó el nivel de ARN m de C4 del paciente con el de dos sujetos normales, usando la transcripción reversa cuantitativa en tiempo real.
Aunque los síntomas continuaron esporádicamente, los datos de laboratorio mejoraron gradualmente sin medicación regular.
En el seguimiento luego de 8 meses luego de su presentación, los niveles de C3 y CH50 fueron normales, y el nivel de C4 se incrementó a 6 mg/dL.
El análisis urinario fue normal.
El componente C4 de la vía clásica del complemento, es una glicoproteína con dos isotipos; C4A y C4B, que se codifican por dos genes situados entre las regiones B y DR del antígeno leucocitario del complejo mayor de histocompatibilidad en el cromosoma 6.
La deficiencia completa de C4 requiere de la presencia de alelos nulos en ambos loci de ambos cromosomas, y es extremadamente raro. Sin embargo, la deficiencia parcial de C4 no es poco común, con porcentajes reportados de C4A y C4B de alelos nulos del 5-15% y 10-20%, respectivamente, en sujetos normales.
Además, varios estudios han mostrado el incremento de la frecuencia de estos alelos nulos en pacientes con SLE y HSP comparado con sujetos normales, sugiriendo que la deficiencia de C4 es uno de los factores mayores de riesgo genéticos para SLE o HSP.
Puede producirse un bajo nivel de complemento por incremento del consumo por la activación de la vía del complemento o por disminución de la producción. Clínicamente, el complemento bajo se diagnostica basado en la combinación de los niveles de C3, C4 y CH50.
En este caso, el nivel de C3 fue normal y el nivel de C4 fue indetectable, con una leve disminución del nivel de CH50, indicando déficit del inhibidor C1 (angioedema hereditario (HAE) o déficit adquirido del inhibidor C1) o déficit de C4.
El HAE y la deficiencia adquirida del inhibidor C1 fueron excluídas porque el nivel del inactivador C1 fue normal y no existían antecedentes familiares. Para la deficiencia C4, aunque no se detectó alelo nulo para C4, el nivel de C4 fue marcadamente bajo a través del período de observación, por lo que se diagnosticó el caso como deficiencia de C4.
Aunque no está claro cuando ocurrió el nivel bajo de C4 en esta paciente, los síntomas iniciales fueron edema leve de los miembros inferiores y púrpura símil HSP desencadenada por un resfrío común a los 44 años de edad. A pesar de los niveles indetectables de C4, los síntomas de la paciente fueron leves y no requirieron tratamiento. Sin embargo, es necesario un seguimiento cuidadoso por la posibilidad de desarrollar SLE, nefritis severa, o algún tipo de enfermedad autoinmune en el futuro.
Los síntomas de déficit de C4 pueden ser muy leves o desapercibidos a pesar del nivel indetectable de C4. En esta paciente, el déficit de C4 se manifestó luego del desarrollo de la púrpura luego de un resfrío común.
Según el conocimiento de los autores, este es el primer reporte de déficit de C4 con expresión de ARNm C4 aumentada.
Tabla 1. Primers usados para transcriptasa reversa PCR de C4.
![]()
¿Qué aporta este artículo a la práctica dermatológica?.
La deficiencia completa de C4 es una condición extremadamente rara. Sin embargo, se ha reportado que la deficiencia parcial de C4 puede ocurrir en sujetos normales, y se asocia con varias enfermedades inmunes. Se reporta el caso de una mujer de 44 años que manifestó edema leve y púrpura en miembros inferiores luego de presentar resfrío.
Presentaba hematuria microscópica persistente y proteinuria, y el nivel de C4 era indetectable. Al exámen histológico de la biopsia, se observó vasculitis leucocitoclásica, con depósito granular de IgG, IgM, C3 y C1q en la paredes de los vasos de dermis superior. La biopsia renal mostró glomerulonefritis proliferativa mesangial leve con leve daño de los capilares, y depósitos granulares de IgM y C4 principalmente en el mesangio.
El nivel de C4 permaneció bajo durante el período de observación, pero no se identificó deficiencia de C4 en el análisis del genotipo y alotipo.
♦ Comentario y resúmen objetivo: Dra. Geraldina Rodríguez Rivello